Hyperparathyroidism
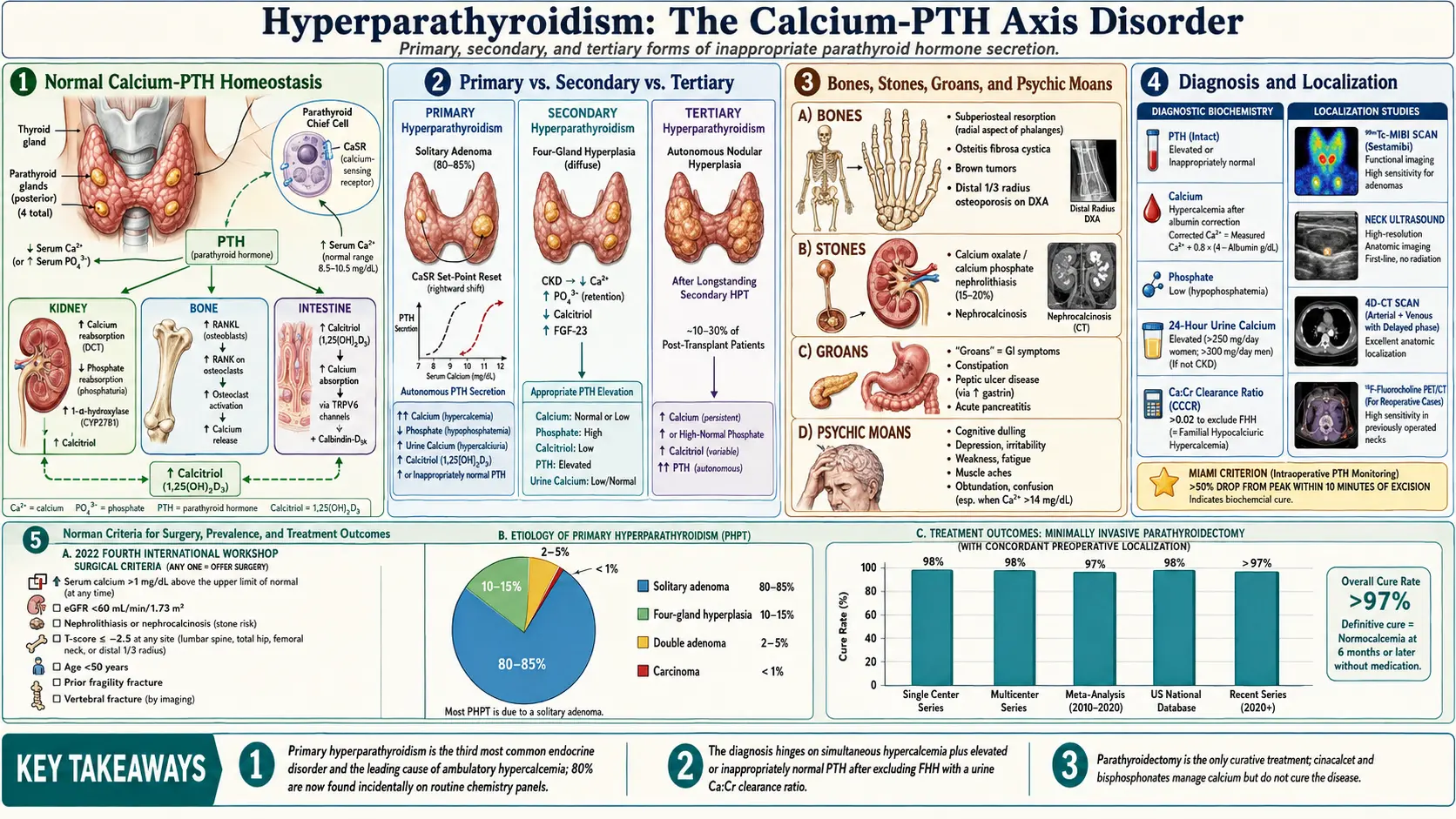
Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Recent Research
- References
- Featured Videos
1. Overview
Hyperparathyroidism is a disorder of calcium homeostasis characterized by inappropriately elevated secretion of parathyroid hormone (PTH) relative to the prevailing serum calcium concentration. It is classified into three principal forms based on pathogenesis and the state of the calcium-PTH feedback axis:
- Primary hyperparathyroidism (PHPT): Autonomous, calcium-independent excess PTH secretion from one or more abnormal parathyroid glands. The calcium-sensing receptor (CaSR) set-point is reset to a higher level, resulting in simultaneous hypercalcemia and inappropriately elevated PTH.
- Secondary hyperparathyroidism (SHPT): Physiologically appropriate compensatory hyperplasia and PTH hypersecretion in response to hypocalcemia, vitamin D deficiency, or phosphate retention — most commonly in the context of chronic kidney disease (CKD). The PTH elevation is appropriate (reactive) and hypercalcemia is absent until late stages.
- Tertiary hyperparathyroidism: Autonomous PTH hypersecretion arising from longstanding SHPT, typically following successful renal transplantation when persistent parathyroid gland hyperplasia produces hypercalcemia despite normalization of the underlying stimulus. Represents a transition from reactive to autonomous PTH secretion.
PHPT is the third most common endocrine disorder after diabetes mellitus and thyroid disease and is the leading cause of hypercalcemia in ambulatory patients. Its clinical spectrum has evolved dramatically since the advent of routine biochemical screening: from a disease of "bones, stones, groans, and psychic moans" to an overwhelmingly asymptomatic condition identified incidentally on multiphasic chemistry panels.
2. Epidemiology
Primary hyperparathyroidism has a population prevalence of approximately 0.1–0.3% (1–3 per 1000 adults), with an annual incidence of approximately 66 cases per 100,000 person-years in community-based studies.
- Sex distribution: PHPT has a striking female predominance, with a female-to-male ratio of 3–4:1. The predominance is most pronounced in postmenopausal women, who account for the majority of cases.
- Age at presentation: Peak incidence occurs in the fifth and sixth decades. However, PHPT can occur at any age, including in children and adolescents (raising concern for hereditary forms).
- Incidental discovery: Approximately 80% of PHPT cases in high-income countries are now diagnosed incidentally on routine laboratory screening, without classic symptoms of hypercalcemia. This proportion was less than 20% prior to the introduction of automated serum chemistry analyzers in the 1970s.
- Post-radiation incidence: Previous therapeutic or inadvertent neck irradiation increases PHPT risk 2–5-fold, with a latency period of 20–50 years.
- Secondary hyperparathyroidism: Virtually universal in patients with advanced CKD (GFR less than 30 mL/min/1.73 m²), affecting essentially 100% of dialysis-dependent patients to varying degrees. Vitamin D deficiency-related SHPT is extremely common globally, given the high prevalence of vitamin D deficiency.
- Normocalcemic PHPT: An emerging entity where PTH is persistently elevated with consistently normal (not merely low-normal) albumin-corrected or ionized calcium, in the absence of secondary causes. Prevalence estimated at 0.4–1.0% in community populations.
3. Pathophysiology
Understanding hyperparathyroidism requires mastery of the calcium-PTH axis and how each form of hyperparathyroidism disrupts it at different points.
Normal Calcium-PTH Homeostasis
Extracellular ionized calcium is maintained within a narrow range (1.15–1.35 mmol/L) through the integrated actions of PTH, calcitriol (1,25-dihydroxyvitamin D), and calcitonin, acting on bone, kidney, and intestine.
The calcium-sensing receptor (CaSR) — a G-protein-coupled receptor of the Family C GPCR superfamily expressed on parathyroid chief cells and renal tubular cells — is the central sensor of ionized calcium. When extracellular Ca²⁺ rises, CaSR activates phospholipase C, generating IP₃ and DAG, which suppress PTH gene transcription, PTH mRNA stability, PTH vesicle exocytosis, and parathyroid chief cell proliferation. Conversely, hypocalcemia releases this inhibition, prompting rapid PTH secretion from pre-formed secretory granules (within seconds to minutes), increased PTH gene expression (within minutes to hours), and parathyroid cellular proliferation (over days to weeks).
PTH exerts its effects through PTH1R, a Gs-coupled receptor:
- Kidney: Stimulates 1α-hydroxylase in proximal tubular cells, converting 25(OH)D to active calcitriol. Increases calcium reabsorption in the distal tubule. Inhibits phosphate reabsorption in the proximal tubule (phosphaturic effect), reducing serum phosphate.
- Bone: Intermittent PTH stimulates osteoblast activity and bone formation (the basis of teriparatide therapy). Continuous PTH excess activates osteoclasts via the RANK-RANKL axis (osteoblasts upregulate RANKL under PTH stimulation), driving net bone resorption, hypercalcemia, and hypercalciuria.
- Intestine: Indirectly increases calcium and phosphate absorption through calcitriol-mediated upregulation of TRPV6 and calbindin-D9k in enterocytes.
Primary Hyperparathyroidism Pathophysiology
In PHPT, the CaSR set-point is reset to a higher level in the abnormal gland(s), requiring a higher calcium concentration to suppress PTH secretion. The sigmoid calcium-PTH response curve is shifted rightward. The result is autonomous PTH secretion producing:
- Hypercalcemia (from enhanced bone resorption and increased intestinal absorption via calcitriol)
- Hypophosphatemia (from PTH-mediated phosphaturia)
- Hypercalciuria (from filtered calcium load exceeding the renal threshold, despite PTH-stimulated distal tubular reabsorption)
- Elevated calcitriol (from PTH-driven 1α-hydroxylase activity in the kidney)
- Subperiosteal bone resorption, osteitis fibrosa cystica (in severe/prolonged disease)
Molecular mechanisms of autonomous PTH secretion include somatic mutations in the MEN1 gene (encoding menin, a tumor suppressor) in up to 20–30% of sporadic adenomas, and cyclin D1 gene rearrangement (with the PTH gene promoter) in 5% — driving unregulated cell proliferation.
Secondary Hyperparathyroidism Pathophysiology
In CKD, SHPT is driven by multiple converging stimuli:
- Hyperphosphatemia: Reduced GFR impairs renal phosphate excretion. Elevated phosphate directly suppresses 1α-hydroxylase and stimulates PTH secretion. Phosphate also stimulates parathyroid cell proliferation. FGF-23, secreted by osteocytes in response to hyperphosphatemia, further suppresses 1α-hydroxylase and increases 24-hydroxylase activity, depleting calcitriol.
- Calcitriol deficiency: Loss of functional nephrons reduces 1α-hydroxylase capacity. Calcitriol normally suppresses PTH gene transcription by binding the vitamin D receptor (VDR) — calcitriol deficiency removes this suppression. Calcitriol also upregulates CaSR expression; its deficiency reduces CaSR density, increasing the set-point for calcium-mediated PTH suppression.
- Hypocalcemia: Calcitriol deficiency reduces intestinal calcium absorption, contributing to hypocalcemia which directly stimulates PTH secretion via CaSR.
- Klotho deficiency: Klotho (a co-receptor for FGF-23) is expressed in the kidney and parathyroid gland. CKD reduces renal Klotho expression, impairing FGF-23 signaling and contributing to FGF-23 resistance and phosphate accumulation. Parathyroid Klotho deficiency impairs direct FGF-23-mediated PTH suppression.
- Parathyroid hyperplasia: Chronic stimulation leads to diffuse hyperplasia, then nodular hyperplasia. Nodular hyperplasia contains monoclonal cell populations with reduced CaSR and VDR expression — transitioning toward autonomous secretion (tertiary HPT).
Tertiary Hyperparathyroidism
After renal transplantation or correction of the underlying stimulus, most patients experience gradual resolution of SHPT. However, 10–30% develop persistent hypercalcemia due to autonomous PTH secretion from hyperplastic glands. The monoclonal nodular hyperplasia that develops in SHPT is associated with somatic loss of CaSR expression and genetic alterations mirroring those in primary adenomas (MEN1 mutations, cyclin D1 overexpression). Gland weight and degree of nodular transformation predict the likelihood of autonomous function persisting after transplant.
4. Etiology and Risk Factors
Primary Hyperparathyroidism
- Solitary parathyroid adenoma: The most common cause (80–85% of PHPT). Typically involves a single gland with monoclonal expansion. The contralateral gland is suppressed. Size ranges from a few millimeters to several centimeters.
- Multigland disease (four-gland hyperplasia): Accounts for 10–15% of PHPT. All four glands are enlarged. More common in hereditary forms (MEN1, MEN2A, HPT-JT). Requires subtotal parathyroidectomy or total parathyroidectomy with autotransplantation.
- Double adenoma: Two separate adenomas in distinct glands; accounts for 2–5% of cases. Intraoperative PTH monitoring is essential to confirm adequate resection.
- Parathyroid carcinoma: Rare (less than 1% of PHPT); should be suspected when calcium is greater than 14 mg/dL, PTH is markedly elevated (often greater than 5× upper limit of normal), and a firm, adherent neck mass is palpable. Associated with mutations in CDC73 (HRPT2), encoding parafibromin.
- Prior neck irradiation: Increases risk of PHPT through radiation-induced parathyroid adenoma formation.
- Lithium therapy: Raises the CaSR set-point for calcium-mediated PTH suppression, producing biochemical changes mimicking PHPT. Prevalence of lithium-induced PHPT is 3–6% of chronically treated patients; removal of the drug may normalize calcium in some.
Hereditary Forms of PHPT
- Multiple Endocrine Neoplasia Type 1 (MEN1): Autosomal dominant; loss-of-function mutations in the MEN1 tumor suppressor gene (chromosome 11q13). PHPT is the earliest and most penetrant manifestation (95% lifetime risk), preceding pancreatic neuroendocrine tumors and pituitary adenomas. Characterized by four-gland hyperplasia and high recurrence rates after surgery. Annual genetic screening recommended for first-degree relatives from age 5.
- Multiple Endocrine Neoplasia Type 2A (MEN2A): Autosomal dominant; gain-of-function mutations in the RET proto-oncogene. PHPT occurs in 20–30% of MEN2A patients (compared to 95–100% for medullary thyroid carcinoma and 50% for pheochromocytoma). Typically mild hypercalcemia.
- Hyperparathyroidism-Jaw Tumor Syndrome (HPT-JT): Autosomal dominant; mutations in CDC73 (HRPT2). Associated with parathyroid adenomas (often cystic), ossifying fibromas of the jaw, renal tumors. Higher risk of parathyroid carcinoma (15–20%).
- Familial Isolated Hyperparathyroidism (FIHP): PHPT without other endocrinopathy. Heterogeneous genetics; some cases harbor MEN1, CDC73, or CASR mutations.
- Familial Hypocalciuric Hypercalcemia (FHH): Autosomal dominant loss-of-function mutations in CASR (type 1), GNA11 (type 2), or AP2S1 (type 3). The CaSR set-point is reset in both the parathyroid gland and the kidney, resulting in mild hypercalcemia, modestly elevated or normal PTH, and paradoxically low urine calcium (calcium-to-creatinine clearance ratio less than 0.01). FHH is benign and does not require surgery — distinguishing it from PHPT is critical to avoid unnecessary parathyroidectomy.
Secondary and Tertiary Hyperparathyroidism Risk Factors
- Chronic kidney disease (the dominant cause of SHPT)
- Vitamin D deficiency (very common; dietary insufficiency, inadequate sun exposure, malabsorption syndromes)
- Malabsorption syndromes: celiac disease, Crohn disease, bariatric surgery, short bowel syndrome
- Pseudo-hypoparathyroidism (target organ resistance to PTH)
- Long-term anticonvulsant therapy (induces CYP450-mediated vitamin D catabolism)
5. Clinical Presentation
Asymptomatic PHPT
The majority of PHPT patients in high-income countries are asymptomatic at diagnosis. Incidental hypercalcemia discovered on routine chemistry panels prompts further workup. These patients may have subtle nonspecific complaints including fatigue, cognitive dulling, mild depression, and constipation that they do not spontaneously report and may not recognize as pathologic until after successful parathyroidectomy.
Classic Symptomatic PHPT
The mnemonic "bones, stones, groans, and psychic moans" encapsulates the classic syndrome of symptomatic PHPT:
"Bones" — skeletal manifestations:
- Osteitis fibrosa cystica: the classic skeletal lesion of severe, longstanding PHPT. Characterized by subperiosteal bone resorption (most prominent on the radial aspect of the middle phalanges of the index finger and middle finger — pathognomonic on radiograph), bone pain, brown tumors (fibrous tissue-filled bone cavities), and "salt-and-pepper" skull on radiograph.
- Diffuse osteopenia and osteoporosis: preferentially affecting cortical bone (distal radius) more than trabecular bone (lumbar spine) in PHPT, contrasting with postmenopausal osteoporosis which primarily affects trabecular sites.
- Subperiosteal resorption, intracortical tunneling, and endosteal scalloping on skeletal radiographs.
- Pathologic fractures in severe disease.
"Stones" — nephrolithiasis and nephrocalcinosis:
- Nephrolithiasis occurs in 15–20% of PHPT patients; the majority are calcium oxalate stones, though calcium phosphate stones are more characteristic. Recurrent stone formers (particularly those with calcium phosphate stones) warrant screening for PHPT.
- Nephrocalcinosis (diffuse calcium deposition in the renal parenchyma) may cause progressive renal insufficiency.
- Presenting features: renal colic, hematuria, urinary tract obstruction.
"Groans" — gastrointestinal manifestations:
- Nausea, vomiting, anorexia, constipation — the most common GI symptoms of hypercalcemia, mediated by reduced smooth muscle contractility.
- Peptic ulcer disease: hypercalcemia stimulates gastrin secretion. Association with Zollinger-Ellison syndrome (gastrinoma) is characteristic of MEN1.
- Acute pancreatitis: classically associated with hypercalcemia above 12 mg/dL. Calcium activates trypsinogen intrapancreatically. Chronic pancreatitis and pancreatic calcifications may occur.
"Psychic moans" — neuropsychiatric manifestations:
- Cognitive impairment, poor concentration, and memory disturbances — often subtle.
- Depression, anxiety, irritability.
- Lethargy, fatigue, generalized weakness.
- Severe hypercalcemia (greater than 14 mg/dL) produces obtundation, stupor, and coma — constituting hypercalcemic crisis.
Hypercalcemic Crisis
A medical emergency characterized by serum calcium typically greater than 14 mg/dL with altered mental status, severe nausea and vomiting, polyuria, polydipsia, and volume depletion. Can be precipitated by dehydration, immobilization, or intercurrent illness. May occur in previously asymptomatic PHPT.
Secondary Hyperparathyroidism (CKD)
- Renal osteodystrophy: a spectrum of skeletal disorders in CKD including osteitis fibrosa cystica (high-turnover, PTH-driven), adynamic bone disease (low-turnover), osteomalacia, and mixed uremic osteodystrophy.
- Vascular and soft tissue calcification: medial calcification of arteries (Mönckeberg's), cardiac valvular calcification, and calciphylaxis (calcific uremic arteriolopathy — painful, ischemic skin lesions with very high mortality).
- Bone pain, fractures, skeletal deformities (particularly in pediatric CKD).
- Pruritus (calcium-phosphate product deposition in skin).
- Cardiovascular morbidity: left ventricular hypertrophy, cardiac fibrosis, arrhythmias.
6. Diagnosis
Initial Biochemical Evaluation
The biochemical hallmark of PHPT is the combination of hypercalcemia with an elevated or inappropriately normal intact PTH (iPTH). The simultaneous interpretation of both values is essential:
- Serum total calcium: Correct for albumin: corrected calcium = measured calcium + 0.8 × (4.0 − albumin [g/dL]). Ionized calcium measurement is more reliable in critically ill patients or those with significant protein abnormalities.
- Intact PTH (iPTH): Measured by two-site immunoradiometric assay (IRMA) or immunochemiluminescent assay (ICMA). In PHPT, PTH is elevated in 85–90% of cases and "inappropriately normal" (within the upper portion of the reference range) in the remaining 10–15%, given that any PTH secretion in the presence of hypercalcemia is inappropriate.
- Serum phosphate: Low-normal or frankly low in PHPT (PTH-induced phosphaturia). Elevated in CKD-related SHPT.
- 24-hour urine calcium: Elevated in most PHPT patients (greater than 400 mg/day). Critical for calculating the calcium-to-creatinine clearance ratio to exclude FHH (ratio less than 0.01 strongly suggests FHH).
- Urine calcium-to-creatinine clearance ratio: (Urine Ca × Serum Cr) / (Serum Ca × Urine Cr). Values greater than 0.02 support PHPT; values less than 0.01 support FHH.
- 25-hydroxyvitamin D: Must be measured to exclude vitamin D deficiency as a cause of secondary PTH elevation or to identify co-existing deficiency complicating PHPT. Vitamin D deficiency should be repleted before interpreting PTH levels.
- Serum alkaline phosphatase (bone-specific): Elevated in high-turnover states; useful for monitoring skeletal disease activity.
- Serum creatinine and eGFR: To assess renal function and exclude CKD as a cause of SHPT.
Differential Diagnosis of Hypercalcemia
Hypercalcemia with elevated PTH essentially confirms PHPT (once FHH is excluded). Hypercalcemia with suppressed PTH (less than 20 pg/mL) indicates a PTH-independent mechanism:
- Malignancy: The most common cause of hypercalcemia in hospitalized patients. Mechanisms include PTHrP secretion (humoral hypercalcemia of malignancy — squamous cell carcinoma, renal cell carcinoma, breast cancer), osteolytic bone metastases (multiple myeloma, breast cancer), and ectopic 1,25(OH)₂D production (lymphomas).
- Granulomatous diseases: Sarcoidosis, tuberculosis, histoplasmosis, berylliosis — autonomous extrarenal 1α-hydroxylase activity in activated macrophages produces excess calcitriol.
- Vitamin D toxicity: Excessive supplementation or granulomatous disease.
- Thiazide diuretics: Reduce renal calcium excretion; mild hypercalcemia; discontinue and re-check before attributing to PHPT.
- Milk-alkali syndrome: Excessive calcium carbonate ingestion; elevated calcium, metabolic alkalosis, renal insufficiency.
- Immobilization: Accelerated bone resorption; most significant in Paget disease, high-turnover states.
- Adrenal insufficiency, hyperthyroidism, acromegaly.
Localization Studies (Preoperative Imaging)
Localization studies are performed only after biochemical diagnosis of PHPT is confirmed — their purpose is surgical planning, not diagnosis. The standard approach combines two complementary modalities:
- Sestamibi (99mTc-MIBI) scintigraphy: Sestamibi preferentially accumulates in mitochondria-rich parathyroid adenomas and is retained longer than in normal thyroid tissue (differential washout). Sensitivity 80–85% for solitary adenomas; lower for multigland disease. May be combined with single-photon emission computed tomography (SPECT) or SPECT/CT for three-dimensional localization.
- Ultrasound of the neck: Sensitivity 70–80% for parathyroid adenomas. Operator-dependent; inferior for ectopic, retrotracheal, or retroesophageal glands. Complements sestamibi well when both localize to the same region (concordant localization), which is optimal before minimally invasive parathyroidectomy.
- Four-dimensional CT (4D-CT): Multiplanar CT with arterial, venous, and delayed phases exploits the hypervascular enhancement pattern of parathyroid adenomas. Sensitivity 70–90%; superior to sestamibi for multigland disease and ectopic locations. Radiation exposure is a consideration in young patients.
- MRI: Useful for persistent/recurrent PHPT after failed surgery and for ectopic mediastinal glands. Sensitivity approximately 70–80% for adenomas.
- Choline PET/CT: Emerging as the highest-sensitivity localization study (greater than 90%) for PHPT, including multigland disease and ectopic glands. Not universally available; may be particularly valuable in reoperative settings. FDA-approved fluorocholine ([¹⁸F]-choline) PET/CT is increasingly endorsed in guidelines.
- Selective venous sampling for PTH: Reserved for reoperative cases when non-invasive localization has failed. Catheterization of neck and mediastinal veins with PTH gradient mapping to lateralize or localize a remnant or missed gland.
Bone Density Assessment
Dual-energy X-ray absorptiometry (DXA) measuring BMD at the lumbar spine, total hip, femoral neck, and distal one-third radius (the cortical site most affected by PHPT) is recommended for all patients with PHPT. The distal radius result is critical — isolated radius osteoporosis at the 1/3 site is a recognized surgical indication.
Norman Criteria for Surgery (Fourth International Workshop 2013, Updated 2022)
The Fourth International Workshop on Asymptomatic PHPT (2013), subsequently updated in guidelines published in 2022, recommends parathyroidectomy when any of the following criteria are met in an otherwise asymptomatic patient:
- Serum calcium: Greater than 1.0 mg/dL (0.25 mmol/L) above the upper limit of normal.
- Renal criteria:
- 24-hour urine calcium greater than 400 mg/day AND increased stone risk by biochemical stone risk analysis (supersaturation indices)
- Nephrolithiasis or nephrocalcinosis by imaging (X-ray, ultrasound, or CT)
- eGFR less than 60 mL/min/1.73 m²
- Skeletal criteria:
- T-score at or below −2.5 at lumbar spine, total hip, femoral neck, or distal one-third radius
- Prior fragility fracture
- Vertebral fracture on imaging (spine X-ray, CT, MRI, or DXA vertebral fracture assessment)
- Age: Less than 50 years (surgery recommended because of longer expected lifespan and disease-related bone loss and complications).
When none of these criteria are met, surgical referral remains a reasonable patient preference, and the decision should be individualized. Observation with monitoring is appropriate for those who decline surgery or have significant operative risk.
Intraoperative PTH Monitoring (Miami Protocol)
Rapid intraoperative PTH (IOPTH) assay — PTH has a half-life of 3–5 minutes — allows real-time confirmation of complete hyperfunctioning gland removal. The Miami Criterion (most widely used): PTH falls by more than 50% from the highest pre-excision value at 10 minutes post-excision AND falls into the normal range. This confirms cure and enables minimally invasive parathyroidectomy under local anesthesia through a small cervical incision, avoiding bilateral neck exploration.
7. Treatment
Surgical Management of Primary Hyperparathyroidism
Parathyroidectomy is the only curative treatment for PHPT and is the treatment of choice for symptomatic patients and asymptomatic patients meeting surgical criteria.
- Minimally invasive parathyroidectomy (MIP): The preferred surgical approach in patients with concordant preoperative localization (sestamibi and ultrasound agreeing on a single adenoma). Involves excision of the localized adenoma through a focused incision under local or general anesthesia, guided by IOPTH monitoring. Cure rates exceed 97% with experienced surgeons. Less surgical morbidity, shorter hospitalization, and better cosmesis compared to bilateral neck exploration.
- Bilateral neck exploration (BNE): The traditional approach, in which all four parathyroid glands are identified and assessed. Required when preoperative localization fails or is discordant, when multigland disease (MEN1, MEN2A) is suspected, or when IOPTH does not meet criteria after single-gland excision. Cure rates: 95–98%.
- Subtotal parathyroidectomy: Removal of 3.5 glands, leaving a well-vascularized half-gland remnant (approximately 50 mg of tissue) in situ. Used for four-gland hyperplasia, particularly in SHPT and MEN1.
- Total parathyroidectomy with autotransplantation: All four glands removed; parathyroid tissue (60–80 mg) is minced and implanted in the non-dominant brachioradialis muscle or subcutaneous forearm fat. Permits easy access for removal if autonomous function develops, without re-entering the neck. Used in MEN1 and severe SHPT.
- Complications of parathyroid surgery:
- Hypocalcemia (transient in 10–30%, permanent in less than 1%): "hungry bone syndrome" — rapid mineral uptake by skeleton after PTH removal; may require prolonged calcium and calcitriol supplementation.
- Recurrent laryngeal nerve injury (transient in 1–5%, permanent in less than 1%).
- Hypoparathyroidism (permanent in less than 1% with single adenoma excision; higher with BNE).
- Cure rates and recurrence: Biochemical cure (normocalcemia) is achieved in 95–99% of operations performed by high-volume surgeons. Recurrence occurs in 1–5% over 10 years for sporadic PHPT; substantially higher (25–50%) in MEN1.
Medical Management of Primary Hyperparathyroidism
For patients who decline surgery, are poor surgical candidates, or do not meet surgical criteria:
- Cinacalcet (calcimimetic): An allosteric positive modulator of the CaSR — increases receptor sensitivity to extracellular calcium, suppressing PTH secretion. Effectively normalizes serum calcium in 70–80% of PHPT patients. Does not improve BMD (unlike surgery). FDA-approved for severe hypercalcemia in PHPT patients for whom parathyroidectomy is not clinically indicated. Dose: 30–90 mg twice daily. Adverse effects: nausea, hypocalcemia.
- Bisphosphonates (alendronate, risedronate, zoledronic acid): Improve BMD at the lumbar spine and femoral neck in PHPT; do not lower serum calcium or PTH significantly. Indicated for patients with osteoporosis who are not surgical candidates. Avoid in patients with eGFR less than 30 mL/min.
- Denosumab: RANKL inhibitor; substantially increases BMD in PHPT. Not FDA-approved for this indication but used off-label; particularly useful in patients with renal impairment precluding bisphosphonates. Rebound hypercalcemia upon discontinuation is a concern.
- Hydration: Adequate oral fluid intake (2–3 L/day) reduces nephrolithiasis risk and maintains renal calcium excretion.
- Dietary calcium: Modest dietary calcium restriction (1000–1200 mg/day) is reasonable; severe restriction paradoxically stimulates PTH secretion and may worsen bone resorption. Dietary calcium does not need to be eliminated.
- Vitamin D replacement: If 25(OH)D is less than 20 ng/mL, gradual repletion with vitamin D3 (800–2000 IU/day) is recommended, with careful calcium and PTH monitoring. Vitamin D deficiency independently stimulates PTH and may worsen the biochemical phenotype.
- Avoidance of thiazide diuretics and lithium when clinically possible, as both raise serum calcium.
Management of Hypercalcemic Crisis
- Aggressive IV saline hydration (300–500 mL/hour initially, then 100–200 mL/hour) to expand volume and promote calciuresis.
- Loop diuretics (furosemide) only after adequate volume repletion to prevent further volume contraction.
- IV bisphosphonate: zoledronic acid 4 mg IV over 15 minutes; onset of calcium-lowering effect in 24–48 hours, peak at 4–7 days.
- Calcitonin: 4 IU/kg SC or IM every 12 hours; rapid onset (4–6 hours) but tachyphylaxis within 48–72 hours due to receptor downregulation. Useful as a bridge while awaiting bisphosphonate effect.
- Denosumab: 60–120 mg SC if bisphosphonate is contraindicated (eGFR less than 30 mL/min).
- Glucocorticoids: effective for vitamin D-mediated hypercalcemia (sarcoidosis, vitamin D toxicity, lymphoma); not effective for PHPT.
- Hemodialysis with low-calcium dialysate: reserved for life-threatening hypercalcemia with severe renal failure.
- Emergency parathyroidectomy: for refractory hypercalcemic crisis with confirmed PHPT and adequate anesthetic risk.
Management of Secondary Hyperparathyroidism in CKD
KDIGO guidelines (2017) target PTH levels of 2–9 times the upper limit of normal in dialysis-dependent patients (to avoid adynamic bone disease from over-suppression).
- Phosphate control: Dietary phosphate restriction; phosphate binders (calcium-based: calcium carbonate, calcium acetate; non-calcium-based: sevelamer, lanthanum carbonate, sucroferric oxyhydroxide) for patients on dialysis or with serum phosphate greater than 4.6 mg/dL in non-dialysis CKD.
- Vitamin D repletion: Nutritional vitamin D (ergocalciferol, cholecalciferol) for patients with 25(OH)D below 30 ng/mL with CKD stages 3–5 not on dialysis.
- Active vitamin D (calcitriol) or vitamin D analogs: Paricalcitol, doxercalciferol — suppress PTH transcription with less risk of hypercalcemia and hyperphosphatemia than calcitriol. Indicated when PTH is persistently elevated above KDIGO targets despite nutrition-focused vitamin D repletion.
- Cinacalcet: CaSR positive allosteric modulator; lowers PTH, calcium, and phosphate in SHPT. The EVOLVE trial failed to show a significant reduction in cardiovascular events in dialysis patients, though post-hoc analyses suggested benefit. Used in combination with vitamin D analogs for refractory SHPT.
- Etelcalcetide: IV calcimimetic administered thrice-weekly with hemodialysis; demonstrated superior PTH suppression versus cinacalcet in head-to-head trials (EVOLVE-2).
- Parathyroidectomy for SHPT: Indicated for severe, refractory SHPT (iPTH persistently greater than 800 pg/mL with hypercalcemia or hyperphosphatemia or both) unresponsive to medical therapy; symptomatic bone disease; or calciphylaxis. Subtotal or total parathyroidectomy with autotransplantation performed.
8. Complications
- Nephrolithiasis and nephrocalcinosis: Present in 15–20% of PHPT; recurrence is high without surgical correction. Chronic nephrocalcinosis leads to CKD.
- Osteoporosis and fractures: PHPT preferentially reduces cortical bone density. Hip and distal radius fracture risk is 2–3-fold elevated. Vertebral fractures occur despite relatively preserved trabecular BMD on DXA (clinical vertebral fracture assessment is essential).
- Cardiovascular disease: Epidemiologic associations include hypertension, left ventricular hypertrophy, endothelial dysfunction, and increased arterial stiffness. Hypertension is present in 40–65% of PHPT patients. Whether these effects are reversed by parathyroidectomy remains controversial; observational data suggest modest blood pressure reduction postoperatively.
- Neurocognitive impairment: Depression, anxiety, cognitive dysfunction, and impaired quality of life are associated with PHPT and may improve after parathyroidectomy, though the evidence for neuropsychiatric benefit is less robust than for nephrolithiasis and bone disease.
- Gastrointestinal complications: Peptic ulceration, pancreatitis. Pancreatitis risk correlates with degree of hypercalcemia.
- Renal insufficiency: Hypercalcemia causes renal vasoconstriction, tubular injury, and nephrocalcinosis — collectively reducing GFR. Severe or prolonged PHPT can produce permanent CKD.
- Hypertensive crisis and arrhythmias: Severe hypercalcemia shortens the QT interval and can precipitate ventricular arrhythmias. Bradycardia and first-degree AV block may occur.
- Calciphylaxis (in SHPT): Life-threatening with mortality up to 60–80%. Characterized by painful, rapidly progressive ischemic skin necrosis from calcium-phosphate deposition in arterioles. Requires urgent intervention including parathyroidectomy in some patients.
- Parathyroid carcinoma: Extremely rare. Aggressive course with local invasion, recurrence, and metastases. Treatment is complete surgical excision; palliative options include cinacalcet for refractory hypercalcemia.
9. Prognosis
- Surgical cure: Parathyroidectomy is curative in 95–99% of sporadic PHPT cases. Post-operatively, serum calcium normalizes within 24–48 hours, and BMD improves significantly over 3–5 years — particularly at the lumbar spine and femoral neck. Renal stones do not recur in the majority. Quality of life improves in most dimensions including energy, mood, and cognitive function.
- Asymptomatic PHPT without surgery: Approximately 25–33% of monitored patients develop indications for surgery within 10 years. Serum calcium is stable in the majority without progressive hypercalcemia. However, subgroup analyses suggest ongoing silent bone loss and neurocognitive decline even in the "stable" group, strengthening the argument for early surgical referral.
- MEN1-related PHPT: High recurrence rate (25–50% over 10 years) despite adequate initial surgery. Lifelong surveillance required. Life expectancy is primarily determined by the associated pancreatic neuroendocrine tumors.
- Secondary/tertiary HPT: Prognosis is largely driven by the underlying renal disease. Adequate PTH control with KDIGO-targeted therapy reduces skeletal morbidity and may reduce cardiovascular calcification progression. Parathyroidectomy for refractory SHPT improves skeletal outcomes and quality of life.
- Parathyroid carcinoma: 5-year survival is approximately 78–85% for completely resected cases; drops to 50% with metastatic disease. Local recurrence is frequent, requiring reoperation.
10. Prevention
- Primary prevention of PHPT: No established preventive strategies for sporadic PHPT. Minimizing neck radiation exposure (shielding during radiographic procedures, limiting therapeutic irradiation to the neck). Genetic counseling for first-degree relatives of patients with MEN1, MEN2A, or HPT-JT.
- Prevention of SHPT: Early correction of vitamin D deficiency in at-risk populations; appropriate dietary phosphate management beginning in CKD stages 3–4; early nephrology referral for progressive CKD to initiate PTH surveillance and timely treatment before advanced SHPT develops.
- Monitoring asymptomatic PHPT: Annual serum calcium, creatinine/eGFR, 24-hour urine calcium; BMD by DXA every 1–2 years; renal imaging (ultrasound or CT) at baseline and as clinically indicated. Spine imaging for vertebral fracture assessment as part of initial and follow-up evaluation.
- Bone protection: Ensure adequate calcium intake (1000–1200 mg/day from dietary sources preferred over supplements), vitamin D sufficiency (25(OH)D greater than 30 ng/mL), weight-bearing exercise, and smoking cessation to minimize skeletal complications in patients under surveillance.
- Genetic screening: Germline genetic testing for MEN1, CDC73, CASR, GNA11, and AP2S1 mutations should be considered in patients with PHPT before age 40, multigland disease, recurrent PHPT, parathyroid carcinoma, jaw tumors, or a family history of hypercalcemia or endocrine neoplasia.
11. Recent Research
- Fluorocholine PET/CT: Multiple prospective studies confirm that [¹⁸F]-choline PET/CT has superior sensitivity (85–95%) compared to sestamibi SPECT/CT for localizing parathyroid adenomas, including multigland disease and ectopic glands. FDA approval (2020) has expanded clinical availability, and updated guidelines now recommend it as a second-line or first-line localization study in reoperative settings.
- Normocalcemic PHPT (NPHPT): The 2022 international workshop guidelines include updated guidance for NPHPT — a phenotype with consistently normal albumin-corrected and ionized calcium but persistently elevated PTH after exclusion of secondary causes. Long-term observational data indicate that 20–40% of NPHPT patients develop hypercalcemia (transitioning to symptomatic PHPT), and skeletal and renal complications parallel those of hypercalcemic PHPT. Surgical indications for NPHPT remain under active investigation.
- Etelcalcetide outcomes in CKD-MBD: The EVOLVE-2 trial and real-world data confirm etelcalcetide superiority over cinacalcet for PTH suppression in hemodialysis patients with SHPT. Cardiovascular and mortality outcome data from long-term registries are maturing.
- Cardiovascular effects of PHPT cure: The OPERA randomized trial (parathyroidectomy versus observation) and several large observational studies have provided more definitive evidence that parathyroidectomy reduces long-term cardiovascular mortality and incident hypertension rates compared to surveillance alone, settling some of the longstanding controversy about cardiovascular benefit.
- Robotics and minimally invasive endoscopy: Robot-assisted transoral endoscopic parathyroid surgery and video-assisted lateral neck approaches are being refined to further minimize cosmetic impact and operative morbidity while maintaining cure rates comparable to traditional MIP.
- Novel calcimimetics and PTH-lowering agents: Upacicalcet (a second-generation IV calcimimetic) and small-molecule CaSR activators are in clinical development for SHPT in CKD. Encaleret, an oral CaSR positive allosteric modulator, is in Phase 3 trials for FHH-related hypercalcemia.
- Bone microarchitecture in PHPT: High-resolution peripheral quantitative CT (HR-pQCT) studies demonstrate trabecular microarchitectural deterioration in PHPT patients with normal DXA BMD, revealing subclinical skeletal disease invisible to conventional DXA. This may refine surgical indications in the future.
12. References
- Bilezikian JP, Khan AA, Silverberg SJ, et al. Evaluation and management of primary hyperparathyroidism: summary statement and guidelines from the fifth international workshop. J Bone Miner Res. 2022;37(11):2293-2314.
- Insogna KL. Primary hyperparathyroidism. N Engl J Med. 2018;379(11):1050-1059.
- Silverberg SJ, Clarke BL, Peacock M, et al. Current issues in the presentation of asymptomatic primary hyperparathyroidism: proceedings of the fourth international workshop. J Clin Endocrinol Metab. 2014;99(10):3580-3594.
- Udelsman R, Åkerström G, Biagini C, et al. The surgical management of asymptomatic primary hyperparathyroidism: proceedings of the fourth international workshop. J Clin Endocrinol Metab. 2014;99(10):3595-3606.
- Eastell R, Brandi ML, Costa AG, et al. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the fourth international workshop. J Clin Endocrinol Metab. 2014;99(10):3570-3579.
- Walker MD, Silverberg SJ. Primary hyperparathyroidism. Nat Rev Endocrinol. 2018;14(2):115-125.
- Marcocci C, Cetani F. Clinical practice. Primary hyperparathyroidism. N Engl J Med. 2011;365(25):2389-2397.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group. KDIGO 2017 clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Suppl. 2017;7(1):1-59.
- Thiem U, Häbel L, Borchhardt K, et al. Management of tertiary hyperparathyroidism after kidney transplantation. Transplant Proc. 2021;53(6):1937-1941.
- Carnaille B. Reoperations for primary hyperparathyroidism: complications and outcome. Langenbecks Arch Surg. 2009;394(5):869-872.
- Kuo EJ, Al-Alusi MA, Du L, et al. Surgery for primary hyperparathyroidism: adherence to consensus guidelines in an academic center. Ann Surg. 2019;269(1):158-163.
- Blanchard C, Mirallié E, Mathonnet M. New insights in primary hyperparathyroidism. Eur Ann Otorhinolaryngol Head Neck Dis. 2012;129(3):140-145.
- Healy P, Robb A, Frilling A, et al. Operative management of parathyroid carcinoma: a series of 34 patients from the National Cancer Data Base. Ann Surg. 2019;269(1):152-157.
- Thanseer N, Bhadada SK, Sood A, et al. Comparative effectiveness of ultrasonography, 99mTc-sestamibi, and 18F-fluorocholine PET/CT in detecting abnormal parathyroid glands in patients with primary hyperparathyroidism. Clin Nucl Med. 2017;42(12):e491-e497.
- Zafereo M, Yu J, Angelos P, et al. American Head and Neck Society Endocrine Surgery Section update on parathyroid localization technology in primary hyperparathyroidism. Head Neck. 2019;41(7):2054-2056.
- Marcocci C, Bollerslev J, Khan AA, Shoback DM. Medical management of primary hyperparathyroidism: proceedings of the fourth international workshop on the management of asymptomatic primary hyperparathyroidism. J Clin Endocrinol Metab. 2014;99(10):3607-3618.
- Block GA, Bushinsky DA, Cheng S, et al. Effect of etelcalcetide vs cinacalcet on serum parathyroid hormone in patients receiving hemodialysis with secondary hyperparathyroidism: a randomized clinical trial. JAMA. 2017;317(2):156-164.
- Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9):2990-3011.
Research Papers
The following PubMed topic searches aggregate the current peer-reviewed literature. Each link opens a live PubMed query — results update as new studies are indexed.
- PubMed — primary hyperparathyroidism
- PubMed — secondary hyperparathyroidism
- PubMed — parathyroid adenoma
- PubMed — parathyroidectomy
- PubMed — hypercalcemia
- PubMed — vitamin D hyperparathyroidism
- PubMed — bisphosphonate hypercalcemia
- PubMed — cinacalcet calcimimetic
- PubMed — sestamibi parathyroid scan
- PubMed — MEN1 MEN2 parathyroid
- PubMed — chronic kidney disease parathyroid
- PubMed — osteoporosis hyperparathyroidism
Connections
- Calcium
- Osteoporosis
- Vitamin D3
- Thyroid Disorders
- Magnesium
- Phosphorus
- Vitamin K
- Kidney Disease
- eGFR
- Creatinine
- Hypertension
- Pancreatitis
- Depression
- Pheochromocytoma
- Sarcoidosis
- Bisphosphonates
- Addison's Disease
- Cushing's Syndrome