Sarcoidosis
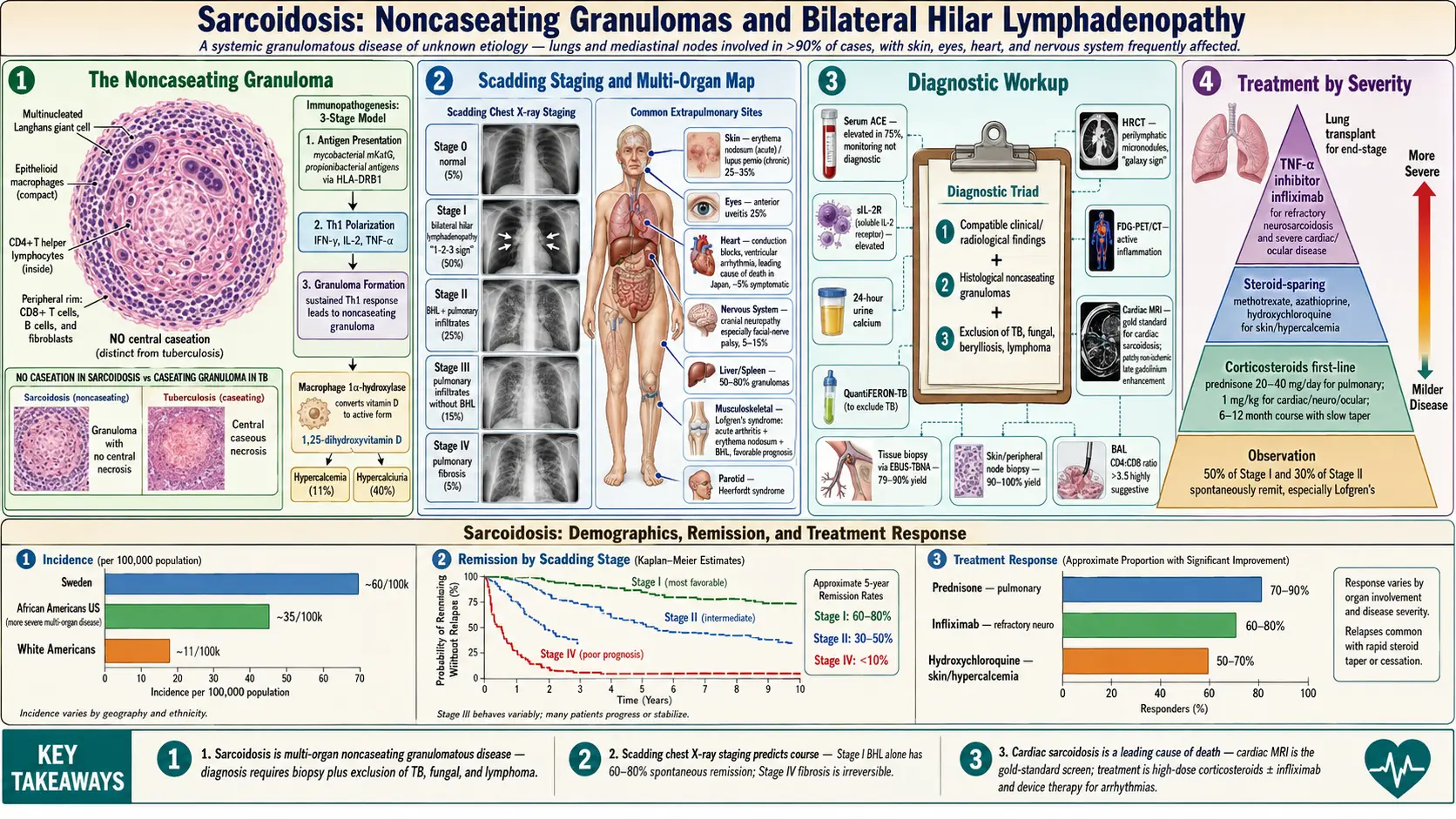
Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Recent Research and Advances
- Research Papers
- Connections
- Featured Videos
1. Overview
Sarcoidosis is a systemic granulomatous disease of unknown etiology, characterized by the accumulation of noncaseating granulomas in affected organs. The lungs and mediastinal lymph nodes are involved in more than 90% of cases, but virtually any organ system can be affected — including skin, eyes, heart, nervous system, liver, spleen, and kidneys. The disease is thought to arise from an exaggerated granulomatous immune response to unidentified environmental antigens in genetically susceptible individuals. Clinical behavior ranges from spontaneous remission to chronic, progressive organ damage. The diagnosis requires compatible clinical and radiological findings, histological demonstration of noncaseating granulomas, and exclusion of other granulomatous diseases.
2. Epidemiology
Sarcoidosis occurs worldwide, with an annual incidence of approximately 10–40 per 100,000 in the United States. The highest incidence is reported in Scandinavian countries (Sweden: ~60 per 100,000) and in African Americans in the United States (incidence ~35 per 100,000, compared to ~11 per 100,000 in White Americans). African Americans also experience more severe, multiorgan disease and higher mortality.
Sarcoidosis predominantly affects young to middle-aged adults, with a peak incidence between ages 25–45 years and a second peak in women aged 50–65. Women are affected slightly more than men overall, but with significant variation by population. The disease is rare in children and those >70 years. The ACCESS study (A Case Control Etiologic Study of Sarcoidosis) established key epidemiological associations, including occupational exposures (insecticide use, microbial bioaerosols) as environmental risk factors.
3. Pathophysiology
The hallmark lesion of sarcoidosis is the noncaseating granuloma — a compact collection of epithelioid macrophages, multinucleated Langhans giant cells, and CD4+ T helper lymphocytes, surrounded by a rim of CD8+ T cells, B cells, and fibroblasts. The absence of central necrosis (caseation) distinguishes sarcoid granulomas from tuberculosis, though overlap can exist.
Immunopathogenesis
Sarcoidosis pathogenesis involves three stages:
- Antigen presentation: Antigen-presenting cells (dendritic cells, alveolar macrophages) encounter putative antigens (mycobacterial proteins — particularly M. tuberculosis catalase-peroxidase (mKatG), propionibacterial antigens, organic dusts). These cells present peptides via HLA class II molecules (notably HLA-DRB1 alleles) to naive CD4+ T cells.
- Th1 immune polarization: Activated CD4+ T cells differentiate predominantly toward Th1 phenotype, releasing IFN-gamma, IL-2, and TNF-alpha. IFN-gamma activates macrophages to form epithelioid cells; TNF-alpha is critical for granuloma formation and maintenance.
- Granuloma formation and evolution: Macrophage clustering and T cell recruitment form the granuloma. Activated macrophages produce 1-alpha-hydroxylase, converting 25-OH vitamin D to 1,25-dihydroxyvitamin D (calcitriol), causing hypercalcemia and hypercalciuria. In resolving sarcoidosis, regulatory T cells (Tregs) suppress the response and granulomas regress. In progressive sarcoidosis, ongoing antigen stimulation and fibroblast activation lead to fibrosis.
Bronchoalveolar lavage classically demonstrates CD4+ lymphocytosis with an elevated CD4:CD8 ratio (>3.5 is highly suggestive of sarcoidosis, though not pathognomonic). The CD4:CD8 ratio reflects the Th1-skewed immune environment. Paradoxically, peripheral blood typically shows relative lymphopenia and anergy.
Specific HLA associations influence disease phenotype: HLA-DRB1*03 predicts acute, resolving disease (Lofgren's syndrome); HLA-DRB1*15 is associated with chronic disease; HLA-DRB1*14 with pulmonary fibrosis.
4. Etiology and Risk Factors
The etiology remains incompletely understood. Current evidence supports a "multiple-hit" model: genetically predisposed individuals develop sarcoidosis upon exposure to specific environmental triggers that drive a dysregulated granulomatous response.
Proposed antigenic triggers:
- Mycobacterial proteins (mKatG, ESAT-6): detected by PCR and mass spectrometry in sarcoid granulomas; no viable organisms identified (rule out active TB)
- Propionibacterium acnes: found in lymph node tissue from Japanese sarcoidosis patients; role uncertain in other populations
- Organic dusts, bioaerosols, and occupational antigens (farmers, firemen)
Genetic risk factors:
- HLA-DRB1 alleles (DRB1*03, *11, *12 associated with disease susceptibility in Europeans)
- Butyrophilin-like 2 (BTNL2) gene variants
- Annexin A11 (ANXA11) variants associated with sarcoidosis susceptibility
- Family history: first-degree relatives have 3–6 fold increased risk
Environmental and occupational exposures:
- Firefighters, construction workers, Navy shipyard workers, nurses (higher incidence)
- World Trade Center responders showed markedly elevated sarcoidosis risk
- Agricultural exposures (animal contact, musty environments)
- Insecticide and pesticide use
Sarcoidosis is not contagious, but rare case clusters (healthcare workers, military personnel) suggest potential infectious or shared environmental triggers.
5. Clinical Presentation
Clinical manifestations depend on the organs involved and the immunological phenotype. Approximately 40–50% of patients are asymptomatic at diagnosis, with incidental findings on chest imaging.
Pulmonary Sarcoidosis (90–95% of cases)
- Dry cough, dyspnea on exertion (most common pulmonary symptoms)
- Chest tightness, wheezing (endobronchial involvement)
- Chest radiograph staging (Scadding classification):
- Stage 0: Normal chest X-ray (5%)
- Stage I: Bilateral hilar lymphadenopathy (BHL) alone (50%)
- Stage II: BHL + pulmonary infiltrates (25%)
- Stage III: Pulmonary infiltrates without BHL (15%)
- Stage IV: Pulmonary fibrosis (5%)
Extrapulmonary Manifestations
- Skin (25–35%): Erythema nodosum (acute; self-limited; good prognosis), lupus pernio (chronic violaceous plaques on nose/cheeks; associated with chronic disease), maculopapular plaques, scar infiltration
- Eyes (25%): Anterior uveitis (most common; acute; presents with photophobia, red eye, pain), posterior uveitis, panuveitis; can cause permanent vision loss if untreated
- Cardiac (5–25% subclinical; ~5% symptomatic): Conduction abnormalities (complete heart block most common; leading cause of sudden death), ventricular arrhythmias, cardiomyopathy (dilated or restrictive), pericarditis; leading cause of sarcoidosis mortality in Japan
- Neurosarcoidosis (5–15%): Cranial neuropathy (facial nerve palsy most common), aseptic meningitis, hypothalamic-pituitary axis dysfunction (diabetes insipidus, hyperprolactinemia), peripheral neuropathy, seizures, mass lesions
- Liver/Spleen (50–80% granulomas at autopsy; 20–30% symptomatic): Hepatomegaly, splenomegaly, elevated alkaline phosphatase; cirrhosis and portal hypertension rare
- Hypercalcemia (11%): Due to macrophage-derived calcitriol; can cause nephrolithiasis, nephrocalcinosis, renal failure
- Musculoskeletal: Arthropathy (Perthes-Jungling cystic bone lesions; Lofgren's syndrome — acute arthritis + erythema nodosum + BHL)
- Parotid gland: Heerfordt syndrome (uveoparotid fever — uveitis + parotid enlargement + fever + facial palsy)
- Upper airways: Nasal mucosal granulomas, hoarseness from laryngeal involvement
6. Diagnosis
No single test is pathognomonic. Diagnosis rests on three pillars: compatible clinical and radiological findings + histological confirmation of noncaseating granulomas + exclusion of alternative diagnoses (especially TB, fungal infections, berylliosis, lymphoma).
Laboratory Evaluation
- Serum ACE: Elevated in ~75% of active sarcoidosis; produced by epithelioid macrophages in granulomas; sensitivity ~60%, specificity ~90%; used for monitoring activity, not diagnosis; also elevated in Gaucher's disease, hyperthyroidism, diabetes
- Serum lysozyme: Elevated in 70% of sarcoidosis; less commonly measured
- Calcium: Hypercalcemia (~11%) and hypercalciuria (~40%); 24-hour urine calcium important to assess renal risk
- 1,25-dihydroxyvitamin D: Elevated; suppressed 25-OH-D3 (consumed in conversion)
- CBC: Lymphopenia, mild anemia; eosinophilia uncommon
- LFTs: Elevated alkaline phosphatase (hepatic involvement)
- QuantiFERON-TB Gold / tuberculin skin test: To exclude TB; typically negative in sarcoidosis (anergy)
- sIL-2R (soluble interleukin-2 receptor): Elevated; better correlation with disease activity than ACE in some studies; particularly useful in neurosarcoidosis and Japanese sarcoidosis
Imaging
- Chest X-ray: Bilateral hilar lymphadenopathy (BHL) is the most characteristic finding; "pawnbroker sign" or "1-2-3 sign" (right paratracheal + bilateral hilar lymphadenopathy). Scadding staging guides prognosis.
- HRCT chest: Perilymphatic micronodules along bronchovascular bundles, subpleural surfaces, and interlobular septa; upper/mid-lobe predominance; "galaxy sign" (cluster of nodules); mediastinal/hilar lymphadenopathy; ground-glass opacity (alveolitis); reticular pattern and traction bronchiectasis (fibrosis in Stage IV)
- FDG-PET/CT: Highly sensitive for active granulomatous inflammation; guides biopsy site selection; evaluates cardiac and systemic involvement; monitors treatment response
- Cardiac MRI: Gold standard for cardiac sarcoidosis; late gadolinium enhancement (patchy, non-ischemic) in ventricular myocardium; evaluates for myocardial inflammation, fibrosis, and functional impairment
- Brain MRI: For neurosarcoidosis; leptomeningeal enhancement, periventricular white matter lesions, hypothalamic/pituitary involvement, cranial nerve thickening
Bronchoscopy and Tissue Biopsy
- Transbronchial lung biopsy (TBLB): Yield 40–90% for pulmonary sarcoidosis (higher in Stage II–III)
- Endobronchial biopsy: Yield ~70% even without visible mucosal lesions (due to submucosal granulomas)
- BAL: CD4:CD8 ratio >3.5 is highly suggestive; BAL lymphocytosis supports diagnosis; not independently diagnostic
- EBUS-TBNA (Endobronchial ultrasound-guided transbronchial needle aspiration): Replaced mediastinoscopy for lymph node sampling; yield 79–90% for Stage I–II sarcoidosis
- Cryobiopsy: Increasing yield compared to conventional TBLB; useful for atypical presentations
- Skin or peripheral lymph node biopsy: Highest yield (90–100%) when accessible lesions are present; should be prioritized
- Conjunctival biopsy: 30% yield even without visible lesions
Echocardiography and Holter Monitoring
Recommended for all sarcoidosis patients given the risk of occult cardiac involvement. Echocardiography detects wall motion abnormalities, cardiomyopathy, and PH. Holter monitoring identifies conduction abnormalities and arrhythmias.
7. Treatment
Not all patients require treatment; ~50% of Stage I and 30% of Stage II patients undergo spontaneous remission. Treatment is indicated for organ-threatening disease, significant symptoms, or progressive functional impairment.
Corticosteroids — First-Line Therapy
- Prednisone 20–40 mg/day (higher doses for cardiac, neurological, and ocular sarcoidosis)
- Initial treatment duration 6–12 months; taper slowly to minimize relapse
- Monitoring: FVC, DLCO, chest X-ray every 3–6 months; serum ACE can guide activity
- Inhaled corticosteroids: Modest benefit for endobronchial sarcoidosis and cough; insufficient for systemic disease
- Topical corticosteroids: Eye drops for anterior uveitis
Steroid-Sparing Agents — Second-Line Therapy
- Methotrexate: 10–15 mg/week; most widely used steroid-sparing agent; effective for pulmonary, cutaneous, ocular, and musculoskeletal sarcoidosis; monthly LFTs; folic acid supplementation; avoid in renal insufficiency
- Azathioprine: 100–150 mg/day; alternative to methotrexate; check TPMT genotype before initiation
- Hydroxychloroquine: Preferred for cutaneous sarcoidosis, hypercalcemia, and fatigue; 200–400 mg/day; annual ophthalmological monitoring
- Leflunomide: 10–20 mg/day; similar efficacy to methotrexate; hepatotoxicity risk
- Mycophenolate mofetil: Used in refractory sarcoidosis; particularly neurosarcoidosis
TNF-Alpha Inhibitors — Third-Line Therapy
- Infliximab: 3–5 mg/kg IV at weeks 0, 2, 6, then every 8 weeks; most evidence-based for refractory sarcoidosis; effective for pulmonary, cardiac, neurological, and cutaneous disease; screen for TB before initiation
- Adalimumab: Alternative to infliximab for subcutaneous self-administration convenience
- Avoid etanercept — not effective in sarcoidosis (soluble TNF receptor decoy; different mechanism from monoclonal antibodies)
Organ-Specific Management
- Cardiac sarcoidosis: High-dose corticosteroids (prednisone 40–60 mg/day); ICD implantation for sustained VT, VF, or high-degree AV block; cardiac transplantation for end-stage cardiomyopathy
- Neurosarcoidosis: High-dose corticosteroids; infliximab for refractory cases; intrathecal methotrexate for CNS involvement
- Hypercalcemia: Corticosteroids; hydroxychloroquine; hydration; avoid vitamin D and sunlight excess; ketoconazole (inhibits 1-alpha-hydroxylase) as adjunct
- Pulmonary fibrosis (Stage IV): Limited evidence for antifibrotics; case reports of nintedanib benefit; lung transplantation for end-stage disease
8. Complications
- Pulmonary fibrosis: Stage IV sarcoidosis; progressive respiratory failure; traction bronchiectasis, bullae, aspergilloma formation in cavities
- Pulmonary hypertension: Multifactorial (hypoxic vasoconstriction, extrinsic compression of pulmonary arteries by lymphadenopathy, vascular granulomas); difficult to treat; worsens prognosis
- Aspergilloma: Aspergillus colonization of fibrotic cavities; hemoptysis (potentially massive); treatment with voriconazole or surgical resection for localized disease
- Sudden cardiac death: Due to ventricular arrhythmia or complete heart block; leading cause of sarcoidosis mortality in some series
- Blindness: From untreated uveitis, glaucoma, or optic nerve involvement
- Renal failure: Nephrocalcinosis and nephrolithiasis from hypercalciuria; interstitial nephritis from granulomatous renal involvement
- Neurosarcoidosis complications: Hydrocephalus, panhypopituitarism, permanent cranial nerve deficits, seizure disorder
- Corticosteroid adverse effects: With long-term treatment — diabetes, osteoporosis, weight gain, adrenal suppression, cataracts
9. Prognosis
The overall prognosis of sarcoidosis is favorable. Approximately 60–70% of patients achieve remission within 2–3 years, either spontaneously or with treatment. Chronic, persistent disease occurs in ~30% and progressive fibrosis in ~10%.
Favorable prognostic indicators:
- Acute onset with erythema nodosum and/or Lofgren's syndrome (spontaneous remission in >90%)
- Stage I disease on chest X-ray (spontaneous remission in ~65%)
- Age <40 at diagnosis
- White ethnicity (milder disease than African Americans)
- HLA-DRB1*03 genotype
Poor prognostic indicators:
- Stage III–IV disease
- Cardiac, neurological involvement
- African American race (more severe, multiorgan disease)
- Lupus pernio (marker of chronic disease)
- Age >40 at onset
- Progressive pulmonary involvement despite treatment
Overall sarcoidosis mortality is approximately 1–7%, primarily from respiratory failure (most common in White patients), cardiac arrhythmias (most common in Japan and African Americans), and neurosarcoidosis.
10. Prevention
There is no established primary prevention strategy for sarcoidosis given its incompletely understood etiology. Secondary prevention focuses on:
- Minimizing occupational exposure to fibrogenic dusts and bioaerosols in high-risk workers
- Early recognition and treatment of ocular involvement to prevent visual loss
- Cardiac monitoring (ECG, Holter, echo) in all diagnosed sarcoidosis patients to detect early arrhythmias and prevent sudden cardiac death
- Bone density monitoring and calcium/vitamin D supplementation (with careful monitoring of serum/urine calcium) during corticosteroid therapy
- Annual ophthalmologic examination for uveitis surveillance
- Avoidance of vitamin D supplements and prolonged sun exposure in patients with hypercalcemia
11. Recent Research and Advances
JAK Inhibitors: Tofacitinib (JAK1/3 inhibitor) and ruxolitinib (JAK1/2 inhibitor) show significant promise in refractory cutaneous sarcoidosis and may have broader pulmonary efficacy. JAK-STAT signaling is activated downstream of key sarcoidosis cytokines (IFN-gamma, IL-6); inhibition disrupts the granulomatous response without the broader immunosuppression of corticosteroids.
Antifibrotic Therapy for Stage IV Sarcoidosis: Small case series and retrospective data suggest nintedanib may slow FVC decline in fibrotic sarcoidosis. Prospective trials are underway; the heterogeneity of pulmonary fibrosis in sarcoidosis (compared to IPF) makes trial design challenging.
Microbiome and Antigen Discovery: Advanced proteomics and mass spectrometry have identified mycobacterial peptides (mKatG, ESAT-6) in sarcoid granulomas across multiple patient populations, strengthening the infectious antigen hypothesis. Whether antibiotic trials targeting Mycobacterium species benefit sarcoidosis patients remains under investigation (CLEAR trial: combination antimycobacterial therapy showed modest benefit).
PET-Guided Management: FDG-PET/CT is increasingly used for response assessment and treatment optimization. PET-CT cardiac sarcoidosis protocols have standardized the evaluation of active cardiac inflammation, enabling more targeted corticosteroid dosing and ICD risk stratification.
Genetic Studies: GWAS have identified novel susceptibility loci including RAB23, OS9, and TNFRSF14. African American-specific risk variants have been identified in ANXA11 and HLA regions, potentially explaining racial disparities in disease severity.
12. References
- Valeyre D, et al. Sarcoidosis. Lancet. 2014;383(9923):1155–1167.
- Judson MA, et al. The WASOG Sarcoidosis Organ Assessment Instrument: An update of a previously validated tool. Sarcoidosis Vasc Diffuse Lung Dis. 2014;31(1):19–27. PMID:24751450
- Hunninghake GW, et al. ATS/ERS/WASOG Statement on Sarcoidosis. Eur Respir J. 1999;14(4):735–737.
- Drent M, et al. Sarcoidosis-associated fatigue. Eur Respir J. 2012;40(1):255–263.
- Baughman RP, et al. Clinical characteristics of patients in a case control study of sarcoidosis (ACCESS). Am J Respir Crit Care Med. 2001;164(10):1885–1889.
- Grutters JC, van den Bosch JMM. Corticosteroid treatment in sarcoidosis. Eur Respir J. 2006;28(3):627–636.
- Baughman RP, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med. 2006;174(7):795–802.
- Hamzeh N, et al. Efficacy of mycophenolate mofetil in sarcoidosis. Respir Med. 2013;107(9):1455–1459.
- Sweiss NJ, et al. HLA-DRB1 is strongly associated with sarcoidosis. PLoS One. 2010;5(11):e15041.
- Crouser ED, et al. Diagnosis and Detection of Sarcoidosis: An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2020;201(8):e26–e51.
- Birnie DH, et al. HRS Expert Consensus Statement on the Diagnosis and Management of Arrhythmias Associated With Cardiac Sarcoidosis. Heart Rhythm. 2014;11(7):1305–1323.
- Herbort CP, et al. International criteria for the diagnosis of ocular sarcoidosis. Int Ophthalmol. 2009;29(6):449–455.
- Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357(21):2153–2165.
- Scadding JG. Prognosis of intrathoracic sarcoidosis in England. Br Med J. 1961;2(5261):1165–1172.
- Morell F, et al. Lymphocyte sensitisation in sarcoidosis. Chest. 1999;116(4):1072–1077.
- Drake WP, et al. (CLEAR). Antimycobacterial therapy effects on T cell responses and disease activity in pulmonary sarcoidosis. Am J Respir Crit Care Med. 2013;187(4):383–389.
Research Papers
The following PubMed topic searches surface the current peer-reviewed literature on Sarcoidosis. Each link opens a live PubMed query; results update as new papers are indexed.
- PubMed search: sarcoidosis
- PubMed search: pulmonary sarcoidosis
- PubMed search: sarcoidosis granuloma
- PubMed search: sarcoidosis corticosteroid treatment
- PubMed search: sarcoidosis methotrexate
- PubMed search: sarcoidosis infliximab TNF
- PubMed search: cardiac sarcoidosis
- PubMed search: neurosarcoidosis
- PubMed search: Lofgren syndrome
- PubMed search: sarcoidosis hypercalcemia vitamin D
- PubMed search: sarcoidosis bronchoalveolar lavage
- PubMed search: sarcoidosis HLA DRB1
Connections
- Pulmonary Hypertension
- Calcium
- Rheumatoid Arthritis
- Interstitial Lung Disease
- Vitamin D3
- Hyperparathyroidism
- Cardiomyopathy
- Tuberculosis
- Lupus
- Diabetes
- Fatigue
- Anemia
- Arrhythmia
- Lymphoma
- Peripheral Neuropathy
- Pneumonia
- Cirrhosis
- Myocarditis