Hemophilia
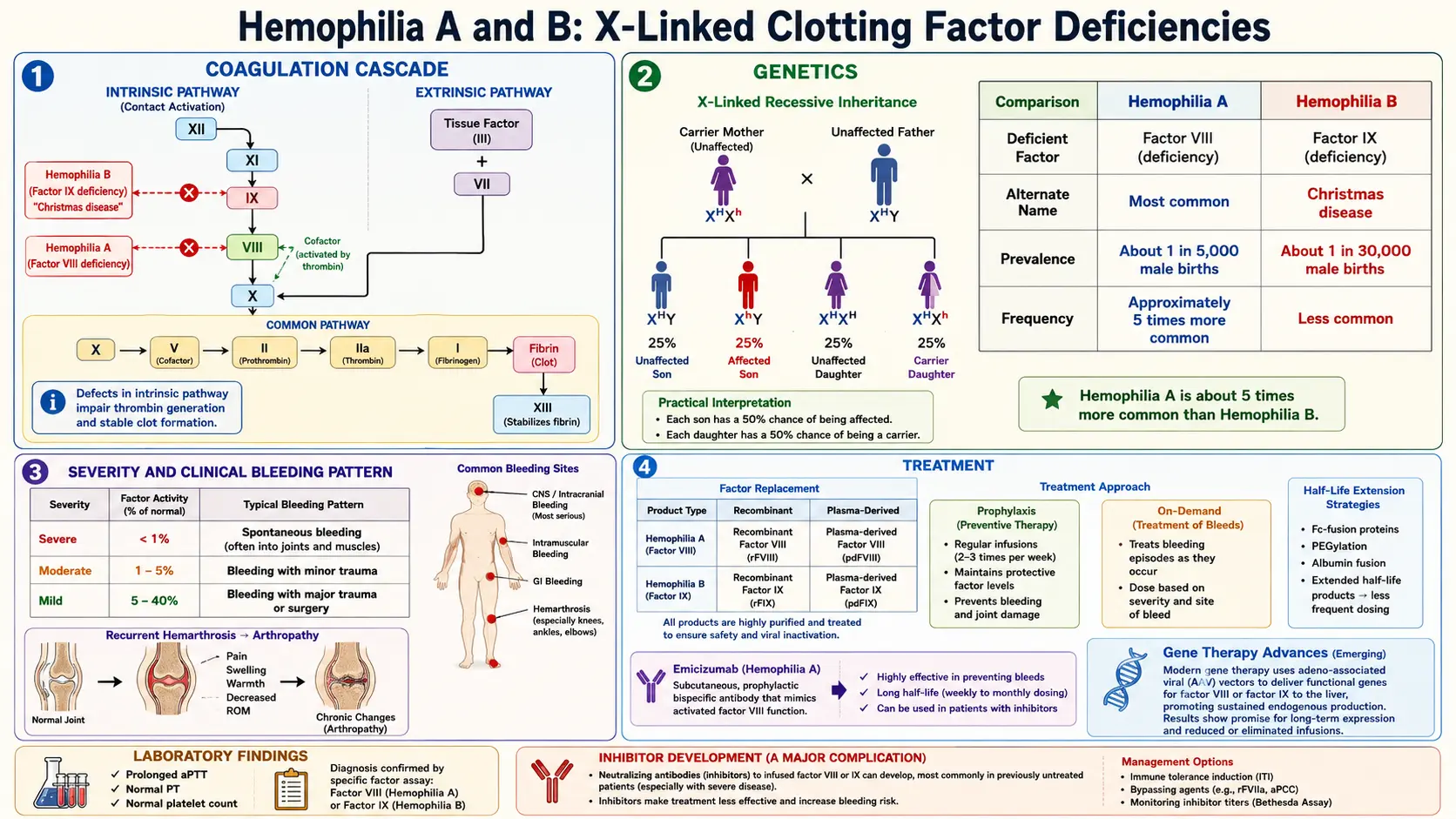
Table of Contents
- Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Recent Research and Advances
- Research Papers
- Connections
- Featured Videos
Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Recent Research and Advances
- References
1. Overview
Hemophilia is a rare, inherited X-linked bleeding disorder characterized by deficiency or dysfunction of specific coagulation factors, resulting in impaired secondary hemostasis and a propensity for prolonged, often spontaneous hemorrhage. The two principal forms are Hemophilia A (Factor VIII deficiency) and Hemophilia B (Factor IX deficiency, also known as Christmas disease). A rarer autosomal form, Hemophilia C (Factor XI deficiency), presents with a milder and more variable bleeding phenotype.
Clinically, hemophilia manifests as excessive bleeding following minor trauma or surgery, and in severe cases, spontaneous bleeding into joints (hemarthrosis), muscles, and internal organs. Without treatment, recurrent hemarthroses lead to progressive joint destruction (hemophilic arthropathy), a major source of morbidity. Modern prophylactic factor replacement therapy has transformed hemophilia from a life-limiting disorder to a manageable chronic condition.
2. Epidemiology
Hemophilia A affects approximately 1 in 5,000 male births worldwide, while Hemophilia B affects approximately 1 in 30,000 male births. Because both conditions are X-linked recessive, they predominantly affect males; females are typically obligate carriers with roughly 50% of normal factor activity, though symptomatic carriers with levels below 40% do occur. The World Federation of Hemophilia (WFH) estimates that approximately 830,000 individuals worldwide have severe hemophilia, though a large proportion remain undiagnosed in low- and middle-income countries.
About 30% of cases arise from de novo mutations without a family history. Severe hemophilia (factor activity <1%) accounts for approximately 43–50% of Hemophilia A cases and 20–45% of Hemophilia B cases. There is no significant racial predilection, and the disorder is distributed globally.
3. Pathophysiology
The Coagulation Cascade
Normal hemostasis involves a tightly regulated cascade of serine protease activations converging on thrombin generation. The intrinsic (contact activation) pathway is initiated by Factor XII activation and proceeds through Factor XI, IX, and VIII. The extrinsic pathway is initiated by tissue factor (TF) complexing with Factor VIIa. Both pathways converge on the common pathway at Factor X activation, leading to prothrombinase complex (Xa + Va) formation, which cleaves prothrombin to thrombin, ultimately converting fibrinogen to fibrin.
In Hemophilia A, deficiency of Factor VIII (FVIII) disrupts the intrinsic tenase complex (FVIIIa–FIXa–phospholipid–Ca²⁺), which normally amplifies Factor X activation by approximately 50-fold compared to Factor VIIa–TF alone. Without adequate FVIII, thrombin generation is markedly reduced, the fibrin clot is fragile, and bleeding persists or recurs as fibrinolysis overwhelms the inadequate clot.
In Hemophilia B, Factor IX (FIX) deficiency similarly impairs the intrinsic tenase complex. FIX is a vitamin K-dependent serine protease; its activated form (FIXa) is the catalytic component of the tenase complex, and without it, Factor X activation through the intrinsic pathway is virtually absent.
Genetic Mechanisms
The F8 gene (encoding FVIII) and F9 gene (encoding FIX) are both located on the long arm of the X chromosome (Xq28 and Xq27.1, respectively). The most common molecular defect causing severe Hemophilia A is an intron 22 inversion, accounting for ~45% of severe cases; intron 1 inversions account for an additional ~5%. Hemophilia B severe cases are caused by a heterogeneous array of point mutations, small deletions, and insertions across the F9 gene. A specific subset, Hemophilia B Leyden, is caused by mutations in the promoter region and characteristically shows spontaneous improvement in factor levels at puberty due to androgen-driven transcription.
The bleeding severity correlates closely with residual factor activity: severe (<1 IU/dL), moderate (1–5 IU/dL), and mild (6–40 IU/dL). Inhibitor development — neutralizing alloantibodies (IgG4) against infused factor concentrates — occurs in ~30% of severe Hemophilia A patients and ~3–5% of Hemophilia B patients, and represents a major therapeutic challenge.
4. Etiology and Risk Factors
- Inherited mutation in F8 gene (Hemophilia A): X-linked recessive; intron 22 inversion most common in severe disease.
- Inherited mutation in F9 gene (Hemophilia B): X-linked recessive; diverse point mutations and deletions.
- De novo mutations: Account for ~30% of cases with no prior family history.
- Female carriers: May be symptomatic if factor activity falls below 40 IU/dL due to skewed X-inactivation (lyonization).
- Acquired hemophilia: Rarely, autoantibodies against FVIII arise spontaneously (acquired hemophilia A) in the setting of autoimmune disease, malignancy, pregnancy, or idiopathically in elderly individuals.
- Inhibitor risk factors: Large deletions or nonsense mutations in F8, African ancestry, intensive early exposure to factor products, and certain HLA haplotypes increase inhibitor risk.
5. Clinical Presentation
Bleeding Patterns
The hallmark of hemophilia is hemarthrosis — spontaneous or traumatic bleeding into joints — which accounts for approximately 75–80% of all bleeding episodes in severe disease. Target joints (most commonly ankles in children, knees and elbows in older patients) undergo repetitive bleeding leading to synovial hypertrophy, cartilage degradation, and hemophilic arthropathy. Muscle hematomas, particularly iliopsoas hematomas, are the second most common bleeding manifestation and can cause femoral nerve compression.
Life-threatening bleeds include intracranial hemorrhage (ICH) (occurring in ~2–8% of patients), gastrointestinal hemorrhage, and retroperitoneal hematomas. Oropharyngeal and neck hematomas can compromise the airway. Even minor procedures — tooth extractions, lumbar punctures, circumcision — can precipitate serious hemorrhage.
Severity Classification (per WFH Guidelines 2020)
- Severe (<1 IU/dL, <1% of normal): Frequent spontaneous bleeds into joints and muscles; typically diagnosed in infancy.
- Moderate (1–5 IU/dL, 1–5%): Occasional spontaneous bleeds; bleeds with minor trauma; diagnosed in early childhood.
- Mild (6–40 IU/dL, 6–40%): Bleeding primarily with significant trauma or surgery; often diagnosed later in life; may be identified incidentally.
6. Diagnosis
Initial Laboratory Evaluation
- Activated partial thromboplastin time (aPTT): Prolonged in Hemophilia A and B; aPTT measures the intrinsic and common pathways.
- Prothrombin time (PT): Normal (extrinsic pathway is intact).
- Platelet count: Normal.
- Thrombin time (TT): Normal.
- Fibrinogen: Normal.
- Bleeding time / PFA-100: Normal (primary hemostasis is intact).
Specific Factor Assays
- Factor VIII activity assay (one-stage or chromogenic): Confirms Hemophilia A; also used to monitor replacement therapy.
- Factor IX activity assay: Confirms Hemophilia B.
- von Willebrand factor antigen (VWF:Ag): Measured to exclude von Willebrand disease type 2N, which can mimic Hemophilia A by reducing FVIII binding.
- Inhibitor screening (Bethesda assay / Nijmegen-Bethesda assay): Performed when there is failure to respond to factor replacement; results expressed in Bethesda Units (BU). Titers ≥5 BU/mL define high-titer (high-responding) inhibitors.
Genetic Testing
Molecular analysis of F8 or F9 confirms diagnosis, identifies the specific mutation, enables carrier testing of female relatives, and informs inhibitor risk stratification. Prenatal diagnosis via chorionic villus sampling or amniocentesis is available for at-risk pregnancies.
7. Treatment
Factor Replacement Therapy
Prophylactic factor replacement is the standard of care for severe hemophilia, aiming to maintain trough factor levels above 1–3% (or higher with extended half-life products) to prevent spontaneous bleeds. The two prophylaxis approaches are:
- Primary prophylaxis: Initiated before age 3 and after the first joint bleed, before joint damage occurs (recommended by WFH).
- Secondary prophylaxis: Initiated after two or more joint bleeds or established joint disease.
Factor concentrates available include plasma-derived concentrates (pathogen-inactivated) and recombinant factor products. Standard half-life (SHL) FVIII has a half-life of ~12 hours (requiring infusion 3 times/week); SHL FIX has a half-life of ~18–24 hours (requiring twice-weekly infusion). Extended half-life (EHL) products incorporate PEGylation, Fc-fusion, or albumin-fusion technologies to extend dosing intervals to once or twice weekly for FVIII and once every 1–2 weeks for FIX.
Non-Factor Therapies
- Emicizumab (Hemlibra): A bispecific monoclonal antibody that bridges FIXa and FX, mimicking the cofactor function of FVIIIa. Administered subcutaneously weekly, biweekly, or monthly. Approved for Hemophilia A with and without inhibitors. Does not correct aPTT and requires specific assays for monitoring.
- DDAVP (desmopressin): Releases endogenous FVIII and VWF from endothelial Weibel-Palade bodies; effective only in mild Hemophilia A; response must be confirmed by pre/post challenge testing.
- Antifibrinolytics: Tranexamic acid (15–25 mg/kg IV/orally TID) and epsilon-aminocaproic acid inhibit plasminogen activators and stabilize clots; used as adjuncts for mucosal bleeds, dental procedures, and menorrhagia.
Inhibitor Management
- Immune tolerance induction (ITI): High-dose FVIII (200 IU/kg/day) administered daily to eradicate inhibitors; success rate ~70% in Hemophilia A.
- Bypassing agents: Activated prothrombin complex concentrate (aPCC; FEIBA) and recombinant FVIIa (NovoSeven) bypass the inhibited factor to generate thrombin via alternative pathways.
- Fitusiran: An RNA interference therapy targeting antithrombin, restoring thrombin generation regardless of factor deficiency or inhibitor status.
- Concizumab: A monoclonal antibody inhibiting tissue factor pathway inhibitor (TFPI), enhancing extrinsic pathway thrombin generation.
Gene Therapy
Adeno-associated viral (AAV) vector-based gene therapy has shown transformative results in clinical trials. Valoctocogene roxaparvovec (Roctavian) for Hemophilia A and etranacogene dezaparvovec (Hemgenix) for Hemophilia B have received regulatory approval, achieving sustained factor expression that eliminates or markedly reduces bleed rates in many recipients. Long-term durability and safety data continue to be accrued.
8. Complications
- Hemophilic arthropathy: Chronic synovitis, cartilage erosion, bone destruction, and joint contractures from recurrent hemarthroses; the leading source of long-term morbidity.
- Intracranial hemorrhage: The leading cause of hemophilia-related mortality; risk is highest in neonates and following trauma.
- Inhibitor development: Complicates ~30% of severe Hemophilia A; greatly increases treatment complexity and cost.
- Pseudotumors: Encapsulated hematomas that can expand and erode adjacent bone; surgical resection often required.
- Bloodborne infection (historical): Prior to viral inactivation of plasma-derived products (pre-1985), widespread HIV and hepatitis C transmission occurred; current products carry negligible infectious risk.
- Anaphylaxis: Particularly a risk with FIX products in Hemophilia B patients with inhibitors (IgE-mediated); nephrotic syndrome has been reported with immune tolerance induction in this population.
9. Prognosis
With modern prophylactic therapy and comprehensive hemophilia treatment center (HTC) care, life expectancy for individuals with hemophilia in high-income countries approaches that of the general population. Patients receiving primary prophylaxis from early childhood can maintain near-normal joint function into adulthood. In low-income settings where factor concentrates are unavailable, severe hemophilia remains life-limiting with significant joint disability. Inhibitor development worsens prognosis by increasing bleed frequency, joint damage, and treatment costs, though immune tolerance induction and emicizumab have substantially improved outcomes. Gene therapy has the potential to provide functional cure for a significant proportion of patients.
10. Prevention
- Genetic counseling: Carrier testing and prenatal diagnosis (chorionic villus sampling, amniocentesis, or preimplantation genetic diagnosis) are offered to at-risk families.
- Primary prophylaxis: Initiated early in severe hemophilia to prevent joint damage before it occurs; the single most effective strategy for preventing arthropathy.
- Comprehensive care: Management at specialized hemophilia treatment centers integrating hematology, orthopedics, physical therapy, dental, and psychosocial support optimizes outcomes and reduces complications.
- Avoidance of NSAIDs and aspirin: These agents impair platelet function and worsen bleeding; acetaminophen is the preferred analgesic.
- Avoidance of intramuscular injections and arterial punctures at non-compressible sites without factor coverage.
- Physical activity: Low-impact exercise (swimming, cycling) strengthens musculature protecting joints while minimizing hemorrhagic risk.
11. Recent Research and Advances
The landscape of hemophilia management is undergoing rapid transformation. Gene therapy has moved from experimental to approved therapy with the licensure of etranacogene dezaparvovec (Hemgenix) for Hemophilia B (2022) and valoctocogene roxaparvovec (Roctavian) for Hemophilia A (2023), with multiple additional AAV and lentiviral constructs in clinical development. Long-term follow-up studies are evaluating durability of expression beyond 5 years.
Fitusiran, an siRNA targeting antithrombin, demonstrated significant bleed reduction in Phase 3 trials (ATLAS program) for both Hemophilia A and B with and without inhibitors, with monthly subcutaneous administration. Concizumab and marstacimab (anti-TFPI antibodies) represent additional non-factor rebalancing strategies. CRISPR-Cas9-based ex vivo correction of F8 and F9 mutations in hepatocytes is under preclinical investigation. Efforts to develop long-acting FVIII molecules beyond current EHL products using von Willebrand factor-decoupling strategies continue to extend dosing intervals. Patient-reported outcome measures and real-world evidence studies are increasingly informing treatment individualization and pharmacokinetic-guided prophylaxis.
Research Papers
The following PubMed topic searches return current peer-reviewed literature relevant to this condition. Each link opens a live PubMed query.
- Hemophilia A factor VIII
- Hemophilia B factor IX
- Hemophilia gene therapy
- Emicizumab hemophilia A
- Hemophilia inhibitors
- Hemophilia prophylaxis
- Hemarthrosis hemophilia
- Hemophilia carrier women
- Hemophilia treatment guidelines
- Extended half-life factor hemophilia
- Desmopressin DDAVP hemophilia
- Hemophilia mortality
Connections
- Von Willebrand Disease
- Thrombocytopenia
- Disseminated Intravascular Coagulation
- Anemia
- Vitamin K
- Iron
- Complete Blood Count
- Deep Vein Thrombosis
- Hemochromatosis
- Polycythemia Vera
- Sickle Cell Disease
- Thalassemia
- Hepatitis C
- Serine
- Side Effects
- Vitamin K and Blood Clotting
- Coagulation Panel