Sickle Cell Disease
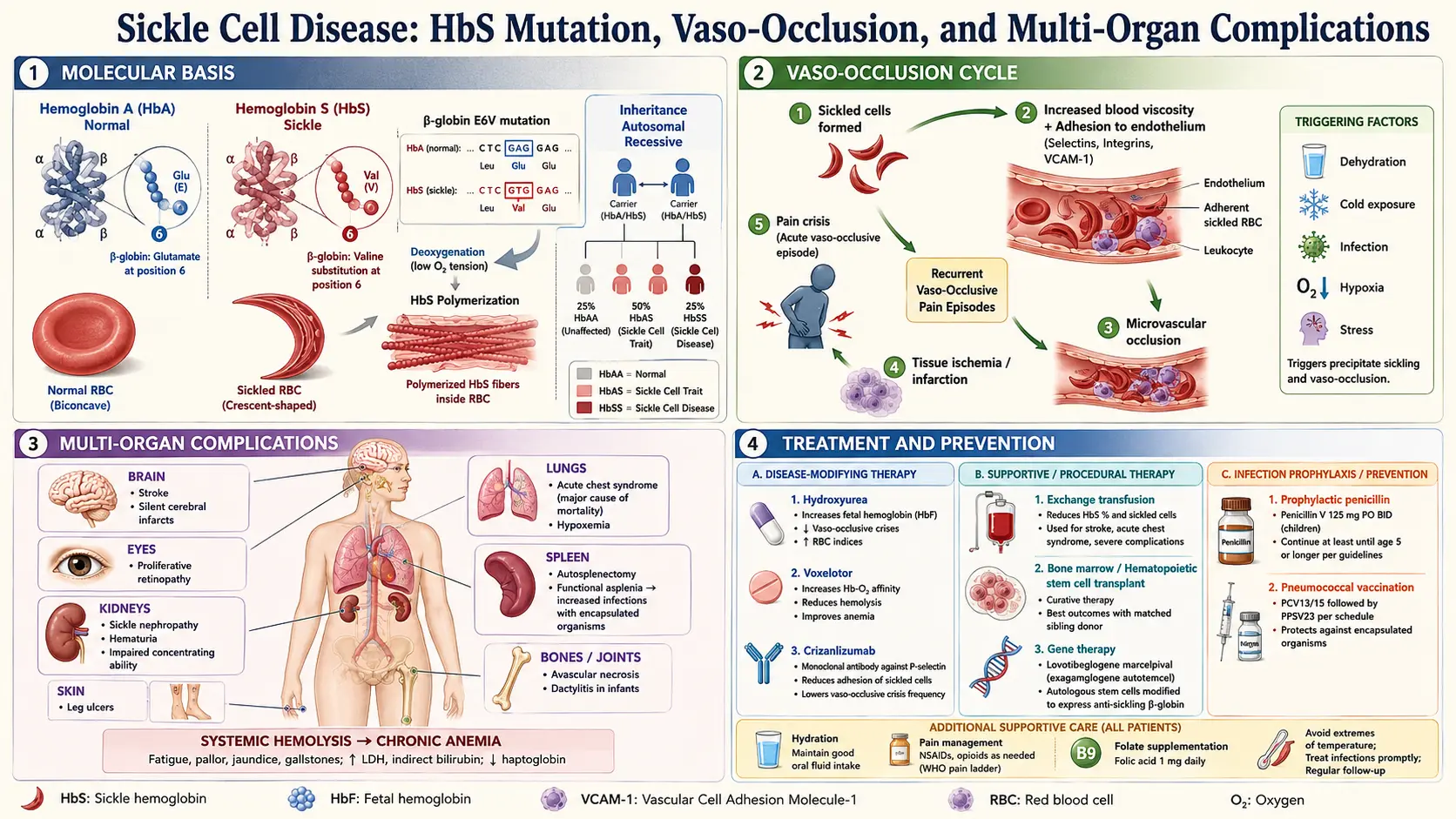
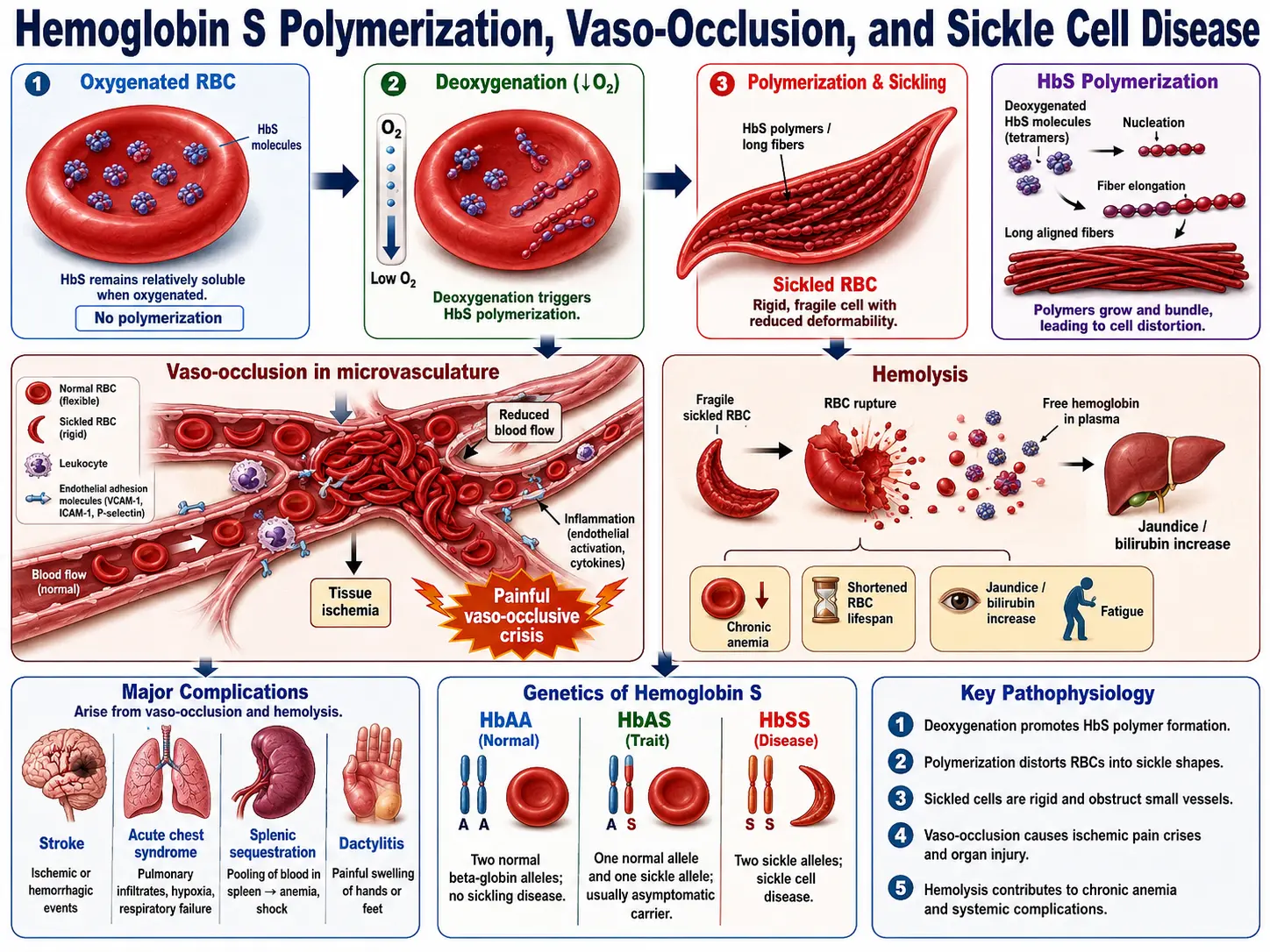
Table of Contents
- Overview
- Genetics and Genotypes
- Pathogenesis: How HbS Causes Disease
- Diagnosis and Newborn Screening
- Complications
- Treatment: Hydroxyurea and Established Drugs
- Gene Therapy and CRISPR
- Supportive Care and Infection Prevention
- Research Papers
- Connections
- Featured Videos
Overview
Sickle Cell Disease (SCD) is the world's most common serious monogenic disorder, affecting approximately 300,000 newborns annually worldwide and 100,000 people in the United States. It is a group of inherited red blood cell disorders caused by a point mutation in the beta-globin gene (HBB) that substitutes glutamic acid for valine at the sixth codon — written as HBB Glu6Val or p.Glu7Val in modern HGVS nomenclature. The resulting abnormal hemoglobin, called hemoglobin S (HbS), behaves normally when oxygenated but polymerizes when deoxygenated, deforming red blood cells into the characteristic rigid sickle shape. These distorted cells obstruct small blood vessels, triggering cycles of ischemia, hemolysis, and inflammation that damage virtually every organ system over a lifetime.
SCD is not a single disease but an umbrella covering several genotypes, all sharing HbS-driven pathology. The most severe form is homozygous HbSS (sickle cell anemia). Before disease-modifying therapy, median survival in high-income countries was approximately 40 years; with hydroxyurea and comprehensive care it now extends into the 50s and 60s, and gene therapy approved in 2023 offers the first broadly accessible curative path.
Genetics and Genotypes
SCD follows autosomal recessive inheritance. Both parents must carry at least one copy of the HbS allele (or a compatible beta-globin variant) for a child to be affected. Carriers — people with sickle cell trait (HbAS) — have one normal and one abnormal allele and are generally healthy, though trait confers rare risk of exercise-induced rhabdomyolysis and renal medullary carcinoma.
Major Genotypes
- HbSS (sickle cell anemia): Two HbS alleles. Most common (roughly 65–70% of SCD in the US) and most severe. HbF (fetal hemoglobin) percentage at birth partially buffers severity and is the target of hydroxyurea therapy.
- HbSC disease: One HbS allele + one HbC allele (HBB Glu6Lys). Milder average severity than HbSS but significant retinopathy and avascular necrosis risk; spleen often preserved → less functional asplenia.
- HbSβ0-thalassemia: HbS + a beta-thalassemia null mutation (no normal HbA produced). Clinically similar to HbSS.
- HbSβ+-thalassemia: HbS + a partial-loss beta-thalassemia mutation (some HbA present). Generally milder than HbSS.
- HbSD, HbSE, HbSOArab: Rare compound heterozygous combinations; severity varies.
Population Distribution
The HbS allele reached high frequency in sub-Saharan Africa, the Mediterranean basin, the Arabian Peninsula, and the Indian subcontinent because HbS heterozygosity confers partial protection against severe Plasmodium falciparum malaria — a striking example of balancing selection. In the US, approximately 1 in 365 African Americans is born with SCD and 1 in 13 carries sickle cell trait.
Pathogenesis: How HbS Causes Disease
Normal adult hemoglobin (HbA, α2β2) stays soluble across physiological oxygen tensions. HbS tetramers, when deoxygenated, polymerize into long intracellular fibers through a nucleation-and-extension process driven by hydrophobic contacts between Val6 on one beta-chain and a hydrophobic pocket on an adjacent molecule. The critical determinant is the oxygen saturation of the red cell and the intracellular HbS concentration — cells with high MCHC (mean corpuscular hemoglobin concentration) and low oxygen tension polymerize fastest.
Vascular Occlusion
Sickled cells are rigid and express abnormal amounts of adhesion molecules (including α4β1 integrin, BCAM/Lutheran, and PS on the outer leaflet). They adhere to activated endothelium expressing E-selectin, VCAM-1, and P-selectin, leading to vaso-occlusion — blockade of post-capillary venules — rather than simple mechanical plugging. Neutrophils and platelets amplify this process. Vaso-occlusion deprives downstream tissue of oxygen, causing ischemic pain (vaso-occlusive crisis, VOC) and, with repeated insults, organ infarction.
Hemolysis
Dense, sickled cells are fragile and destroyed in the microcirculation and spleen. The resulting intravascular and extravascular hemolysis releases free hemoglobin and heme into plasma. Free hemoglobin scavenges nitric oxide (NO), the principal vasodilator and platelet inhibitor, creating a NO-deficient state that promotes vasoconstriction, platelet activation, and pulmonary hypertension. Hemolysis elevates unconjugated bilirubin (causing jaundice and gallstones), depletes haptoglobin, and raises LDH — the lab signature of active hemolysis.
Endothelial Inflammation
Free heme activates Toll-like receptor 4 (TLR4) on endothelial cells, triggering NF-κB-dependent upregulation of adhesion molecules and pro-inflammatory cytokines. This creates a self-amplifying cycle: sickling → hemolysis → NO depletion + TLR4 activation → endothelial inflammation → adhesion molecule expression → more sickling and vascular occlusion.
Diagnosis and Newborn Screening
Early diagnosis is critical because SCD complications begin in infancy. Neonatal sepsis from encapsulated organisms and splenic sequestration can be fatal before age 2 without prophylaxis, making newborn screening a life-saving public health intervention.
Newborn Screening
All 50 US states mandate newborn screening for SCD using isoelectric focusing (IEF), high-performance liquid chromatography (HPLC), or hemoglobin electrophoresis on the dried blood spot collected at 24–48 hours of life. The pattern "FS" (fetal hemoglobin + HbS, no HbA) indicates HbSS or HbSβ0-thalassemia; "FSC" indicates HbSC. A confirmatory test is performed at 2 months of age.
Diagnostic Workup in Older Patients
- HPLC or hemoglobin electrophoresis: Quantifies each hemoglobin fraction (HbS, HbA, HbF, HbC, etc.) — the definitive diagnostic test.
- Complete blood count: Normocytic or microcytic anemia (Hgb typically 6–9 g/dL in HbSS), reticulocytosis (5–15%), elevated WBC (reactive leukocytosis common), thrombocytosis.
- Hemolysis markers: Elevated LDH and indirect bilirubin, low haptoglobin, elevated reticulocyte count.
- Peripheral smear: Sickle cells, target cells, polychromasia, and Howell-Jolly bodies (reflecting functional asplenia).
- Transcranial Doppler (TCD): Annual screening in children aged 2–16 years for elevated cerebral blood flow velocities indicating stroke risk (≥200 cm/s in the middle cerebral or internal carotid artery = high risk).
- Molecular/genetic testing: DNA analysis for HBB genotype, particularly to distinguish HbSβ0 from HbSβ+ thalassemia, or for atypical presentations.
Complications
SCD affects every organ system. A few complications dominate morbidity and mortality.
Vaso-Occlusive Crisis (VOC)
The most frequent acute complication. Severe pain — usually in the back, chest, extremities, or abdomen — caused by ischemia from microvascular occlusion. Precipitants include cold, dehydration, hypoxia, stress, and infection. Management: IV fluids, opioid analgesia (individualized dosing), NSAIDs (if renal function permits), and supplemental oxygen for hypoxia. Hospitalization often required. Chronic VOC leads to cumulative organ damage.
Acute Chest Syndrome (ACS)
The leading cause of death in SCD. Defined as a new pulmonary infiltrate on chest imaging + at least one of: chest pain, fever, respiratory symptoms, or oxygen desaturation. Causes include fat embolism from infarcted bone marrow, infection (respiratory viruses, Mycoplasma, Chlamydia), and in-situ sickling in the pulmonary microvasculature. Treatment: supplemental oxygen, incentive spirometry, empiric antibiotics (a macrolide or fluoroquinolone for atypicals), simple or exchange transfusion for severe cases, and bronchodilators.
Stroke
Children with SCD have an 11% risk of overt stroke by age 20 without prophylaxis — 200-fold higher than the general pediatric population. Large-vessel arteriopathy (stenosis/occlusion of the intracranial internal carotid and proximal middle cerebral arteries) is the mechanism, distinct from the adult stroke mechanism. TCD screening identifies children with blood flow velocities ≥200 cm/s who benefit from regular blood transfusion therapy (reduces stroke risk from ~10% to ~1% per year). Adults face silent cerebral infarcts and hemorrhagic stroke from moyamoya-type collateral vessels.
Splenic Sequestration
Acute massive enlargement of the spleen due to trapping of sickled red cells. Causes rapid anemia, thrombocytopenia, and circulatory collapse. Predominantly affects children (the spleen infarcts and atrophies by late childhood in HbSS — "autosplenectomy") and some HbSC patients who retain a functional spleen longer. Treatment: urgent transfusion. Risk of recurrence is high; splenectomy considered after a severe episode.
Aplastic Crisis
Transient arrest of erythropoiesis caused by parvovirus B19 infection. The virus selectively infects erythroid precursors. In normal individuals, a week of marrow suppression is tolerated; in SCD patients with a red cell lifespan of only 10–20 days (vs. 120 days normal), even brief marrow aplasia causes precipitous anemia. Reticulocyte count drops to near zero. Usually self-limited over 10–14 days; transfusion support may be needed.
Functional Asplenia and Infection Risk
Repeated splenic infarction causes progressive loss of splenic function, leaving patients vulnerable to encapsulated organisms: Streptococcus pneumoniae, Haemophilus influenzae type b, and Neisseria meningitidis. Overwhelming post-splenectomy infection (OPSI) can be rapidly fatal. Prevention: penicillin prophylaxis from age 2 months to 5 years (and often beyond); immunization with conjugate pneumococcal, Hib, meningococcal, and annual influenza vaccines.
Priapism
Ischemic (low-flow) priapism — painful, unwanted erection persisting >4 hours — occurs in 30–45% of males with HbSS by age 20. Caused by NO depletion and sluggish blood flow in corpora cavernosa. Recurrent episodes (stuttering priapism) lead to erectile dysfunction. Acute management: IV fluids, opioids, aspiration of corporal blood, intracavernous sympathomimetics (phenylephrine); exchange transfusion for prolonged episodes. Hydroxyurea reduces recurrence.
Avascular Necrosis (AVN)
Ischemic death of bone, most commonly the femoral and humeral heads, from vascular occlusion in the terminal nutrient arteries supplying subchondral bone. Cumulative prevalence exceeds 50% by the 4th decade. Pain, limited range of motion, and ultimately joint collapse requiring arthroplasty.
Chronic Organ Damage
Kidneys (glomerulopathy, papillary necrosis, renal failure), eyes (proliferative retinopathy → vitreous hemorrhage and retinal detachment, especially in HbSC), lungs (pulmonary hypertension in ~10%, associated with worst prognosis), liver (congestive hepatopathy, intrahepatic cholestasis), and heart (cardiomegaly, diastolic dysfunction) all sustain cumulative injury over decades.
Treatment: Hydroxyurea and Established Drugs
Hydroxyurea (Hydroxycarbamide)
Hydroxyurea remains the foundational disease-modifying therapy, FDA-approved for adults in 1998 and children in 2017. It elevates fetal hemoglobin (HbF) levels by reactivating the gamma-globin genes — HbF dilutes intracellular HbS, raising the concentration threshold for polymerization. Hydroxyurea also reduces neutrophil count and endothelial adhesion molecule expression, directly dampening vascular inflammation. The landmark MSH trial (PMID 8667871) demonstrated a 44% reduction in VOC frequency and a 50% reduction in ACS episodes. Long-term follow-up data show a survival benefit of roughly 40% at 26 years for patients who take it consistently. Starting dose is typically 15–20 mg/kg/day, titrated to the maximum tolerated dose (MTD) targeting mild myelosuppression (ANC 2,000–4,000/µL).
L-Glutamine (Endari)
FDA-approved in July 2017 for patients ≥5 years with SCD. L-glutamine is the precursor for NAD synthesis in red blood cells; SCD red cells have reduced NAD redox potential, making them more susceptible to oxidative hemolysis. A phase 3 trial (PMID 29363597) showed that oral L-glutamine (0.3 g/kg twice daily, max 30 g/day) reduced acute sickle cell crises by 25% and hospitalizations by 33% versus placebo. Mechanism: replenishing NADH and NADPH buffers oxidative damage during hypoxia.
Crizanlizumab (Adakveo)
FDA-approved November 2019 for patients ≥16 years. A humanized monoclonal antibody targeting P-selectin, the adhesion molecule expressed on activated platelets and endothelium that mediates white cell and red cell adhesion to the vessel wall. The SUSTAIN trial (PMID 27959701) showed a 45.3% reduction in median annual VOC rate versus placebo (1.63 vs. 2.98 crises/year). Administered as monthly IV infusions (5 or 10 mg/kg). Can be combined with hydroxyurea.
Voxelotor (Oxbryta)
FDA-approved November 2019 for patients ≥12 years (later expanded to ≥4 years). Voxelotor binds covalently to the alpha-chain of hemoglobin, stabilizing the oxygenated (R-state) conformation and directly inhibiting HbS polymerization. The HOPE trial (PMID 31869785) demonstrated a 73% response rate in hemoglobin improvement (≥1 g/dL rise) versus 17% placebo, and significant reductions in hemolysis markers. Dose: 1,500 mg orally once daily. Note: In November 2024, the FDA requested voluntary market withdrawal after a post-approval study suggested a possible increase in VOC and deaths; clinical decisions should follow current FDA guidance and clinical judgment.
Chronic Transfusion Therapy
Regular red cell transfusions (simple or exchange) dilute HbS with donor HbA, reducing the fraction of circulating sickle cells. Indications include: primary and secondary stroke prevention (especially in children with high TCD velocities), recurrent ACS, and end-stage organ failure bridging to transplant. Risks: iron overload (requiring chelation with deferasirox or deferoxamine), alloimmunization (30–50% of chronically transfused SCD patients develop alloantibodies), and infection transmission. Exchange transfusion (erythrocytapheresis) removes sickle cells while adding donor cells, limiting iron accumulation.
Gene Therapy and CRISPR
After decades of development, two gene therapies received FDA approval in December 2023, representing the first curative options broadly accessible beyond matched sibling HSCT.
Betibeglogene Spartacus (Zynteglo) — Lentiviral Gene Addition
Approved by the FDA on August 17, 2023 for patients ≥12 years with HbSS or HbSβ0-thalassemia who have a history of VOC. Betibeglogene spartacus is a lentiviral vector (BB305) that inserts a modified beta-globin gene (encoding anti-sickling βT87Q hemoglobin) into the patient's own hematopoietic stem cells ex vivo. Autologous CD34+ cells are collected via apheresis, transduced, then reinfused after myeloablative busulfan conditioning. The HGB-206 study (PMID 35255496) showed that 93% of patients with SCD achieved freedom from severe VOC, with durable hemoglobin responses. A single-course treatment with a list price of $3.1 million — outcomes-based rebate contracts negotiated with payers.
Exagamglogene Autotemcel (Casgevy) — CRISPR-Cas9 Gene Editing
Approved by the FDA on December 8, 2023 — the world's first approved CRISPR-based medicine — for patients ≥12 years with HbSS or HbSβ0-thalassemia who have recurrent VOC. Casgevy uses CRISPR-Cas9 to disrupt the BCL11A enhancer in erythroid cells, derepressing the fetal hemoglobin loci (HBG1/HBG2) and reactivating HbF production. It does not require a healthy donor beta-globin sequence — it exploits the fact that HbF tetramers (α2γ2) do not participate in HbS polymerization. The CLIMB-SCD-121 trial (PMID 37924528) demonstrated that 28 of 29 evaluable patients were free from severe VOC for at least 12 consecutive months after treatment. Because it edits the patient's own cells, there is no graft-versus-host disease risk. The price is approximately $2.2 million. Both CRISPR and lentiviral therapies require myeloablative conditioning — eligibility is limited by fitness for chemotherapy.
Allogeneic Hematopoietic Stem Cell Transplant (HSCT)
The long-established curative option. Best results with a matched sibling donor: disease-free survival approximately 90–95% in pediatric patients. Matched unrelated donor transplants carry higher risks of graft failure and graft-versus-host disease, though outcomes have improved with better HLA typing and reduced-intensity conditioning. HSCT is standard of care for children with severe disease who have an available matched sibling donor; gene therapy expands curative options to patients without a donor.
Supportive Care and Infection Prevention
Comprehensive supportive care substantially improves outcomes and quality of life even without curative therapy.
- Penicillin prophylaxis: Twice-daily oral penicillin V (or amoxicillin) from age 2 months through at least 5 years reduces pneumococcal sepsis by over 80% (PMID 3897864). Many centers continue indefinitely or until pneumococcal vaccination series is complete.
- Vaccinations: Conjugate pneumococcal (PCV15 or PCV20), polysaccharide pneumococcal (PPSV23), Hib, meningococcal (MenACWY + MenB), hepatitis B, and annual influenza vaccines are all recommended.
- Hydration: Patients should maintain good oral hydration; dehydration increases MCHC and sickling risk.
- Folic acid supplementation: 1 mg/day orally compensates for accelerated red cell turnover and increased folate demand.
- Pain management: A written individualized pain management plan (including home-based opioid dosing for VOC) reduces ED visits and hospitalization. NSAIDs are effective adjuncts if renal function permits.
- Ophthalmology surveillance: Annual retinal exams starting at age 10 for HbSS, earlier for HbSC (higher retinopathy prevalence).
- Avoidance of triggers: Extreme cold, altitude without acclimatization, dehydration, and heavy exertion without adequate warm-up are recognized VOC triggers.
- Psychosocial support: Chronic pain, frequent hospitalizations, and functional limitations make depression and anxiety common comorbidities. Mental health support and social work coordination are integral to comprehensive SCD care.
Research Papers
The following peer-reviewed publications represent landmark and recent research on sickle cell disease pathogenesis, treatment, and gene therapy.
- Charache et al. (1995) MSH trial: hydroxyurea in adults with sickle cell anemia — PMID 8667871
- Niihara et al. (2018) L-glutamine oral therapy for sickle cell disease — PMID 29363597
- Ataga et al. (2017) SUSTAIN trial: crizanlizumab for sickle cell disease — PMID 27959701
- Vichinsky et al. (2019) HOPE trial: voxelotor in sickle cell disease — PMID 31869785
- Kanter et al. (2022) HGB-206 study: betibeglogene spartacus gene therapy — PMID 35255496
- Frangoul et al. (2024) CRISPR exagamglogene autotemcel (Casgevy) for SCD — PMID 37924528
- Gaston et al. (1986) Prophylaxis with penicillin in sickle cell anemia (PROPS trial) — PMID 3897864
- Adams et al. (1998) TCD-guided transfusion to prevent stroke in sickle cell disease (STOP trial) — PMID 9462351
- Wang et al. (2008) Hydroxycarbamide in very young children with sickle-cell anaemia (BABY HUG) — PMID 18768946
- Platt et al. (2013) Mortality in sickle cell disease: life expectancy and risk factors — PMID 23601682
- Ware et al. (2015) Hydroxycarbamide versus chronic transfusion in prevention of recurrent stroke (TWiTCH) — PMID 25651877
- Kato et al. (2018) Sickle cell disease: a global clinical priority — PMID 29515624
Connections
- Anemia
- Thalassemia
- Stroke
- Hemophilia
- Thrombotic Thrombocytopenic Purpura
- Hemolytic Uremic Syndrome
- Iron
- Folate
- Complete Blood Count
- Heart Failure
- Fatigue
- Jaundice
- Pulmonary Hypertension
- Chest Pain
- Chronic Pain
- Thrombocytopenia
- Disseminated Intravascular Coagulation
- Kidney Stones