Thrombocytopenia
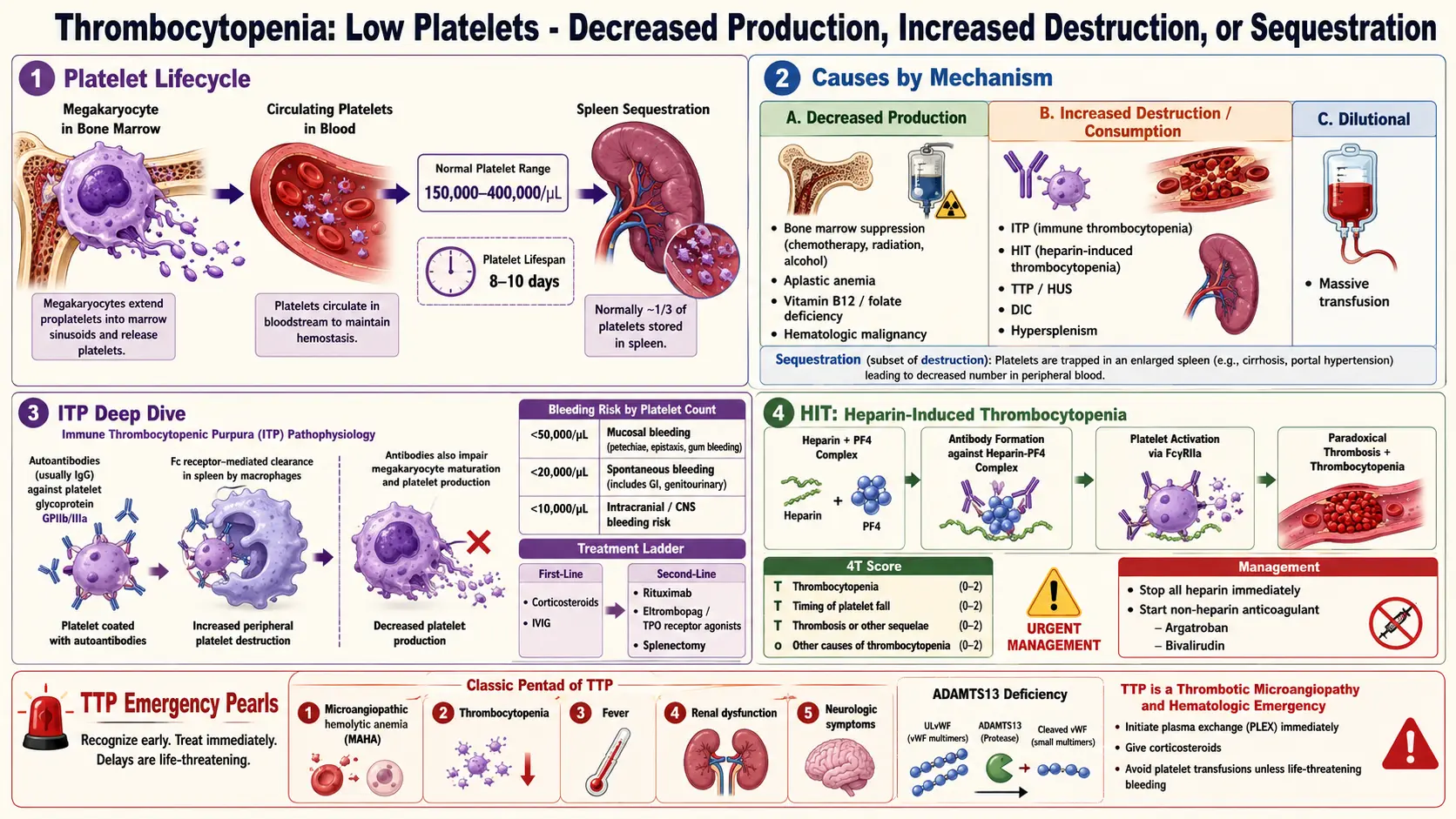
Table of Contents
- Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Recent Research and Advances
- Research Papers
- Connections
- Featured Videos
Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Recent Research and Advances
- References
1. Overview
Thrombocytopenia is defined as a platelet count below the lower limit of normal, conventionally set at 150 × 10⁹/L (150,000/µL). Platelets are anucleate cell fragments derived from bone marrow megakaryocytes that play a fundamental role in primary hemostasis — adhering to sites of vascular injury, activating, and aggregating to form the initial platelet plug that is subsequently reinforced by the fibrin clot of secondary hemostasis.
Thrombocytopenia can result from decreased platelet production, increased platelet destruction or consumption, platelet sequestration, or dilution. The clinical significance ranges from incidental, asymptomatic mild thrombocytopenia (platelet count 100–149 × 10⁹/L) to life-threatening hemorrhage in severe thrombocytopenia (<20 × 10⁹/L). The most clinically important etiologies include immune thrombocytopenia (ITP), heparin-induced thrombocytopenia (HIT), and thrombotic thrombocytopenic purpura (TTP), each with distinct pathophysiology and management.
2. Epidemiology
Thrombocytopenia is common across clinical settings. In hospitalized patients, it is estimated to occur in 25–41% of patients, most often related to sepsis, medications, or critical illness. Immune thrombocytopenia (ITP) has an incidence of approximately 2–10 cases per 100,000 adults per year; the pediatric form is more common (4–8 per 100,000 children per year) and usually follows a viral illness with spontaneous resolution in 80–90% of cases. In adults, ITP is more common in women of childbearing age (female-to-male ratio ~3:1), though this sex predilection diminishes with age.
Gestational thrombocytopenia (mild, incidental thrombocytopenia of pregnancy) occurs in approximately 5–8% of all pregnancies and accounts for 70–80% of thrombocytopenia in pregnancy. Heparin-induced thrombocytopenia (HIT) occurs in 0.1–5% of patients receiving unfractionated heparin, depending on patient population and duration of exposure. Drug-induced thrombocytopenia from other agents (e.g., quinine, vancomycin, valproic acid) has an overall incidence of ~10 cases per million persons per year.
3. Pathophysiology
Platelet Production and Destruction
Platelet production occurs in bone marrow through megakaryopoiesis: hematopoietic stem cells differentiate under the influence of thrombopoietin (TPO) (produced primarily by the liver) into megakaryocyte progenitors, mature megakaryocytes, and ultimately into platelets through a process of cytoplasmic fragmentation (thrombopoiesis). Each megakaryocyte generates 1,000–3,000 platelets. Platelets circulate for approximately 7–10 days before being cleared by the reticuloendothelial system (spleen and liver).
Thrombocytopenia arises through three principal mechanisms:
- Decreased production: Bone marrow failure (aplastic anemia), infiltration (leukemia, lymphoma, metastatic cancer, myelofibrosis), ineffective thrombopoiesis (B12/folate deficiency, myelodysplastic syndrome), chemotherapy/radiation-induced suppression, viral infections (HIV, hepatitis C, EBV).
- Increased destruction or consumption:
- Immune-mediated: ITP — autoantibodies (predominantly IgG) bind platelet surface glycoproteins (GPIIb/IIIa, GPIb/IX), leading to accelerated Fc-receptor-mediated phagocytosis by splenic macrophages. CD8+ T cells also contribute to megakaryocyte and platelet destruction. In HIT, IgG antibodies against complexes of heparin and platelet factor 4 (PF4) bind platelet FcγRIIA receptors, causing platelet activation, consumption, and paradoxically thrombosis.
- Non-immune: Mechanical destruction in microangiopathic hemolytic anemias (TTP, HUS, DIC); consumptive coagulopathy.
- Sequestration: Splenomegaly (from cirrhosis, portal hypertension, myeloproliferative neoplasms) sequesters up to 90% of the total platelet pool (normally ~33%), reducing circulating platelet count.
Coagulation Impact
Platelets contribute to secondary hemostasis by providing a phospholipid surface for assembly of the prothrombinase complex (FXa–FVa) and the tenase complex (FIXa–FVIIIa), amplifying thrombin generation approximately 300,000-fold compared to reactions in solution. Severe thrombocytopenia thus impairs both primary hemostasis and the amplification of secondary hemostasis, explaining the propensity for mucocutaneous and internal bleeding.
4. Etiology and Risk Factors
- Immune thrombocytopenia (ITP): Primary (idiopathic) or secondary to SLE, antiphospholipid syndrome, CLL, HIV, hepatitis C, H. pylori infection.
- Heparin-induced thrombocytopenia (HIT): Unfractionated heparin (UFH) > low-molecular-weight heparin (LMWH); higher risk in surgical/orthopedic patients, women.
- Drug-induced thrombocytopenia (DITP): Quinine, quinidine, vancomycin, linezolid, valproic acid, carbamazepine, checkpoint inhibitors, GPIIb/IIIa inhibitors (abciximab, tirofiban).
- Thrombotic microangiopathies: TTP (ADAMTS13 deficiency), HUS (Shiga-toxin or complement-mediated), DIC, HELLP syndrome.
- Bone marrow disorders: Aplastic anemia, MDS, leukemia, myeloma, myelofibrosis, metastatic solid tumor infiltration.
- Nutritional deficiencies: Vitamin B12 and folate deficiency (megaloblastic marrow, ineffective megakaryopoiesis).
- Infections: Sepsis, HIV, hepatitis C, EBV, CMV, dengue fever, malaria, leptospirosis.
- Hypersplenism: Cirrhosis, portal hypertension, storage diseases, myeloproliferative neoplasms.
- Gestational thrombocytopenia: Benign, incidental; platelet count rarely <70 × 10⁹/L; resolves postpartum.
- Congenital thrombocytopenias: Bernard-Soulier syndrome, Wiskott-Aldrich syndrome, MYH9-related disorders, thrombocytopenia-absent radius (TAR) syndrome.
5. Clinical Presentation
Bleeding Patterns by Severity
- Platelet count 100–150 × 10⁹/L: Usually asymptomatic; no significant bleeding risk in the absence of other hemostatic defects.
- Platelet count 50–100 × 10⁹/L: Minimal bleeding risk; excessive bleeding may occur with major trauma or surgery.
- Platelet count 20–50 × 10⁹/L: Increased bruising and mucocutaneous bleeding; surgical procedures require platelet support.
- Platelet count <20 × 10⁹/L: Significant risk of spontaneous bleeding; petechiae, purpura, epistaxis, gingival bleeding, menorrhagia.
- Platelet count <10 × 10⁹/L: High risk of life-threatening hemorrhage including intracranial hemorrhage.
Clinical Features
- Petechiae: Non-blanching pinpoint hemorrhages in the skin, particularly on dependent areas and mucous membranes; highly characteristic of platelet-type (primary hemostasis) bleeding.
- Purpura: Confluent areas of intradermal hemorrhage; "wet purpura" in the oral mucosa indicates severe thrombocytopenia.
- Ecchymoses: Large, flat bruises disproportionate to trauma.
- Mucosal bleeding: Epistaxis, gingival oozing, menorrhagia, GI bleeding, hematuria.
- HIT-specific findings: Paradoxical thrombosis (arterial or venous) in the setting of falling platelet count 5–14 days after heparin initiation; skin necrosis at heparin injection sites; anaphylaxis with heparin bolus.
6. Diagnosis
Initial Workup
- Complete blood count with differential and peripheral blood smear: Confirm thrombocytopenia; evaluate for platelet clumping (EDTA-dependent pseudothrombocytopenia — repeat with citrate tube), giant platelets (congenital macrothrombocytopenias), schistocytes (TMA), blasts, or hypersegmented neutrophils (B12/folate deficiency).
- PT and aPTT: Normal in ITP; prolonged in DIC, liver disease, vitamin K deficiency; aPTT prolonged in antiphospholipid syndrome.
- Fibrinogen and D-dimer: Hypofibrinogenemia and elevated D-dimer suggest DIC or TMA.
- LDH and haptoglobin: Elevated LDH and absent haptoglobin suggest hemolysis (TTP, HUS).
- Reticulocyte count: Elevated in hemolytic processes.
- ADAMTS13 activity and inhibitor: Severely reduced (<10%) with inhibitor confirms TTP.
- HIT testing: 4T score for pre-test probability; PF4-heparin ELISA (sensitive, not specific); serotonin release assay (SRA; gold standard for confirmation).
- Antiplatelet antibody testing: Limited sensitivity and specificity; not routinely recommended for ITP diagnosis; ITP is a diagnosis of exclusion.
- Bone marrow aspirate and biopsy: Indicated when marrow failure or infiltration is suspected; shows increased megakaryocytes in ITP (reactive) vs. hypocellularity (aplastic anemia) or dysplastic megakaryocytes (MDS).
- H. pylori testing: Urea breath test or stool antigen test recommended in all adult ITP patients.
- HIV and hepatitis C serology: Standard workup for secondary ITP.
7. Treatment
Immune Thrombocytopenia (ITP)
- First-line therapy: Corticosteroids — prednisone 1 mg/kg/day for 2–4 weeks, then taper; or dexamethasone 40 mg/day for 4 days (pulsed). Response in 60–80%; durable remission in only ~30%. Intravenous immunoglobulin (IVIG) 1 g/kg/day × 1–2 days: Rapidly elevates platelet count (within 24–48 hours) via Fc receptor blockade and altered platelet clearance; preferred in severe bleeding or urgent procedures. Anti-D immunoglobulin (in Rh-positive, non-splenectomized patients) is an alternative to IVIG.
- Second-line therapy: Thrombopoietin receptor agonists (TPO-RAs) — romiplostim (subcutaneous weekly; Nplate) and eltrombopag (oral daily; Promacta) stimulate megakaryopoiesis by binding the TPO receptor (c-Mpl); response rates ~80%; sustained responses off therapy occur in ~30%. Rituximab (anti-CD20; 375 mg/m² weekly × 4): Depletes B-lymphocytes producing autoantibodies; overall response ~60%, durable response ~20%.
- Splenectomy: 60–70% long-term remission rate; deferred unless refractory to medical therapy due to surgical risk and asplenia complications; laparoscopic approach preferred.
- Fostamatinib (SYK inhibitor): FDA-approved for chronic ITP; inhibits Fc receptor signaling in macrophages, reducing platelet phagocytosis.
- Avatrombopag: An oral TPO-RA approved for chronic ITP and thrombocytopenia in chronic liver disease.
HIT Management
- Immediate cessation of all heparin (including flushes, heparin-coated catheters) upon clinical suspicion.
- Alternative anticoagulation with a non-heparin agent: Argatroban (direct thrombin inhibitor; preferred in renal impairment — hepatically cleared) or bivalirudin; fondaparinux (Factor Xa inhibitor) is widely used off-label. DOACs (rivaroxaban, apixaban) are increasingly used for ongoing anticoagulation after the acute phase.
- Avoid platelet transfusions (may worsen thrombosis by providing substrate for HIT antibody-mediated activation, though used in life-threatening bleeding).
- Warfarin is contraindicated in acute HIT until platelet count recovery (>150 × 10⁹/L) due to risk of limb gangrene from protein C depletion.
Platelet Transfusion Thresholds
- Active major bleeding: Transfuse to maintain platelets >50 × 10⁹/L (or >100 × 10⁹/L for CNS or ophthalmic bleeding).
- Prophylactic: Major surgery: maintain >50 × 10⁹/L; lumbar puncture, epidural, or minor procedure: >50 × 10⁹/L; central line insertion: >20 × 10⁹/L; stable non-bleeding: >10 × 10⁹/L.
- Avoid transfusion in TTP and HIT unless there is life-threatening hemorrhage.
8. Complications
- Intracranial hemorrhage: The most feared complication; risk rises dramatically below 10–20 × 10⁹/L; associated with ~40% mortality in ITP.
- HIT-associated thrombosis: Deep vein thrombosis, pulmonary embolism, arterial thrombosis (limb, coronary, cerebral); limb gangrene; associated with up to 20–30% mortality if untreated.
- Chronic ITP: Disease persisting beyond 12 months (>3 months = persistent); associated with significantly impaired quality of life, infection risk from immunosuppression, and treatment-related side effects.
- Post-splenectomy sepsis: Life-threatening overwhelming infection with encapsulated organisms (Streptococcus pneumoniae, Haemophilus influenzae, Neisseria meningitidis).
- GI hemorrhage: Potentially life-threatening in severe thrombocytopenia.
9. Prognosis
Prognosis depends critically on the underlying etiology. Pediatric ITP is self-limiting in ~80–90% of cases within 6–12 months. Adult ITP is more often chronic; approximately 20–30% achieve durable remission with first-line therapy, while many require long-term second-line treatment. With TPO-RAs, most patients maintain sustained platelet responses, though long-term treatment may be necessary. HIT, if recognized and managed promptly with alternative anticoagulation, carries a good prognosis; unrecognized or inadequately treated HIT is associated with a 30-day thrombosis rate of ~50%. Thrombocytopenia secondary to bone marrow failure (aplastic anemia, leukemia, MDS) carries the prognosis of the underlying disease. TTP has a mortality rate of >90% untreated but falls to 10–20% with plasma exchange.
10. Prevention
- HIT prevention: Use LMWH rather than UFH whenever clinically appropriate (lower HIT incidence with LMWH); minimize duration of heparin exposure; use alternatives to heparin (fondaparinux, bivalirudin) in heparin-allergic or HIT-history patients.
- Drug-induced thrombocytopenia prevention: Review medications in any patient with thrombocytopenia; avoid re-exposure to implicated agents; document drug allergies prominently.
- Platelet transfusion refractoriness prevention: Use ABO-compatible, leukoreduced products; consider HLA-matched platelets in alloimmunized patients.
- Infection-related thrombocytopenia prevention: Antimicrobial stewardship and prompt treatment of sepsis.
- H. pylori eradication: Achieves platelet count recovery in ~50% of H. pylori-positive ITP patients, potentially obviating immunosuppressive therapy.
11. Recent Research and Advances
The field of ITP has been transformed by thrombopoietin receptor agonists, with romiplostim and eltrombopag now standard second-line options. Newer TPO-RAs — avatrombopag and lusutrombopag — have expanded the oral treatment armamentarium. Fostamatinib, an SYK inhibitor, represents a mechanistically distinct approach targeting macrophage Fc receptor signaling. The anti-neonatal Fc receptor (FcRn) inhibitor efgartigimod alfa (Vyvgart) has shown efficacy in Phase 3 ITP trials (ADAPT+), rapidly reducing pathogenic IgG antibodies including anti-platelet autoantibodies, offering a novel mechanism distinct from all existing therapies.
For HIT, real-world data increasingly support direct oral anticoagulants (particularly rivaroxaban and apixaban) for treatment and secondary prevention, avoiding the complexity of parenteral non-heparin anticoagulants. Better understanding of anti-PF4 antibody biology — including the recognition of spontaneous, non-heparin-triggered HIT-like syndromes — has refined serologic testing and management of immune-mediated thrombocytopenia more broadly. Caplacizumab (anti-VWF nanobody) in combination with plasma exchange and immunosuppression has become standard of care for TTP, reducing TTP-related mortality and relapse.
Research Papers
The following PubMed topic searches return current peer-reviewed literature relevant to this condition. Each link opens a live PubMed query.
- Immune thrombocytopenia ITP
- Heparin-induced thrombocytopenia HIT
- Thrombotic thrombocytopenic purpura TTP
- Thrombocytopenia pregnancy
- Gestational thrombocytopenia
- Thrombopoietin receptor agonist thrombocytopenia
- Romiplostim eltrombopag ITP
- Platelet transfusion threshold
- Drug-induced thrombocytopenia
- Thrombocytopenia guidelines
- Bone marrow failure thrombocytopenia
- Neonatal alloimmune thrombocytopenia
Connections
- Anemia
- Deep Vein Thrombosis
- Vitamin K
- Complete Blood Count
- Hemophilia
- Von Willebrand Disease
- Polycythemia Vera
- Disseminated Intravascular Coagulation
- Sepsis
- Hepatitis C
- Leukemia
- Cirrhosis
- Liver Disease
- Hemochromatosis
- Sickle Cell Disease
- Preeclampsia
- Pulmonary Embolism
- Tick Borne Co Infections
- Mononucleosis