Iron Overload & Hidden Iron Toxicity
Few claims in functional nutrition are as counter-intuitive as the one Morley Robbins makes about iron. Mainstream medicine and the lay public share the same mental model: iron is a scarce, hard-to-absorb nutrient; low ferritin or hemoglobin is “low iron”; the answer is to take more. The Root Cause Protocol turns that on its head. Robbins argues that the typical American adult is not iron-deficient at all — the typical American adult is iron-loaded in the wrong tissues while the iron in the right tissues fails to function because ceruloplasmin’s ferroxidase activity has collapsed. Adding more iron makes the picture worse, not better. This article walks through that thesis — the iron paradox, the difference between bound and free iron, what ferritin actually measures, the role of mandatory food fortification, the spectrum of hereditary hemochromatosis, brain iron in neurodegeneration, why blood donation is a therapy and not just a charitable act, when iron supplementation is genuinely warranted, and the practical RCP off-loading protocol.
Table of Contents
- The Iron Paradox: Tired Yet Iron-Loaded
- Free Iron vs. Bound Iron
- Ferritin: Storage Marker or Inflammation Marker?
- The Iron-Fortified American Food Supply
- Hereditary Hemochromatosis & The Spectrum
- Brain Iron Accumulation in Neurodegeneration
- Therapeutic Phlebotomy: Donating Blood as Medicine
- When Iron Supplementation IS Genuinely Needed
- Practical Approach to Iron Off-Loading
- Key Research Papers
- Connections
- Featured Videos
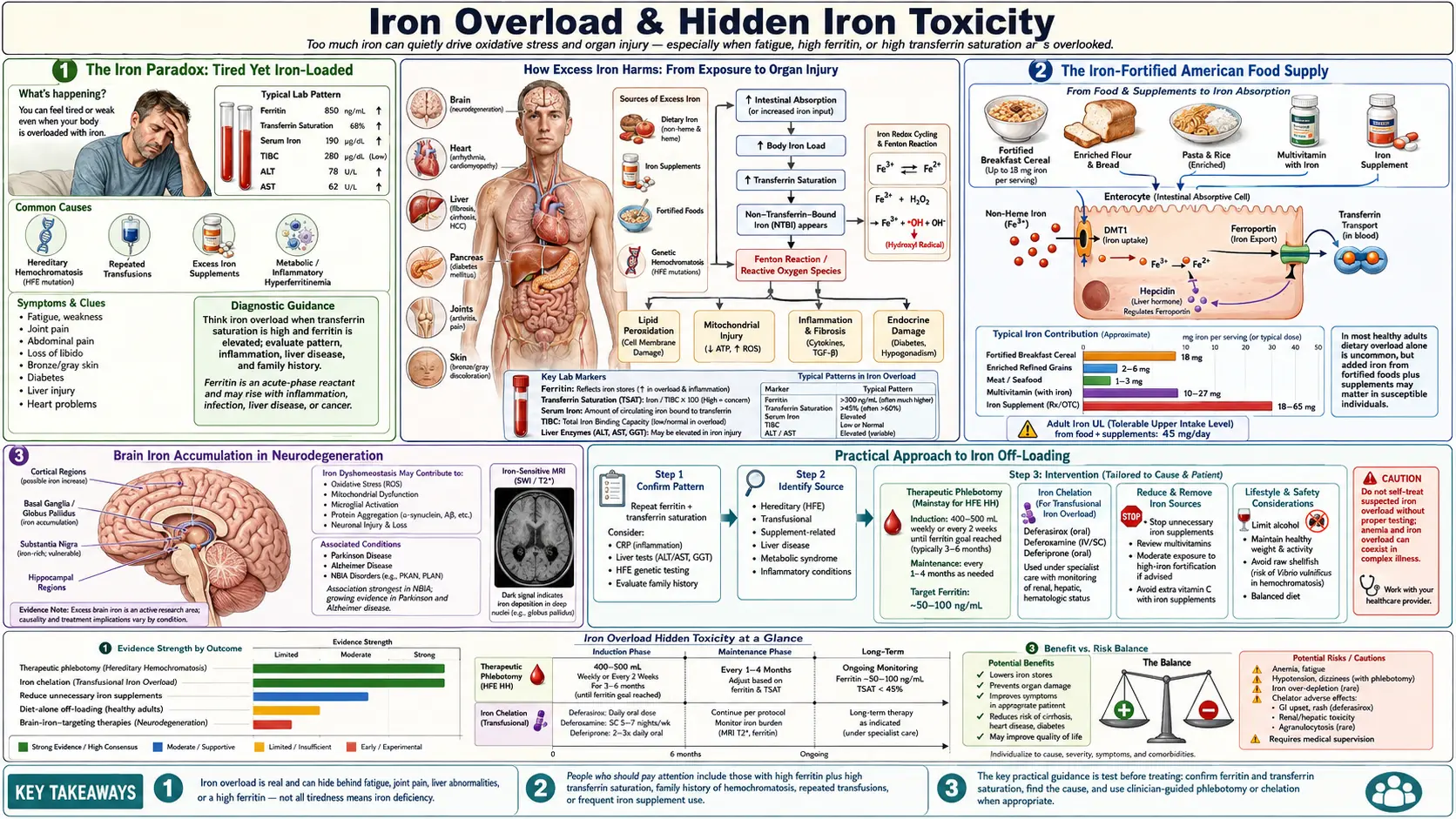
1. The Iron Paradox: Tired Yet Iron-Loaded
Robbins’s central observation is one most clinicians can confirm if they look. A patient walks in exhausted. Hemoglobin is 11.8 g/dL — flagged low. Ferritin is 22 ng/mL — flagged low. The standard lab printout pushes both numbers into the “iron-deficient” column, and the standard prescription is ferrous sulfate or a multivitamin with 18 mg of iron. The patient takes it for six months. Hemoglobin barely moves. The fatigue gets worse. GI symptoms appear. Inflammatory markers (CRP, ferritin itself) climb. Eventually a liver MRI is ordered for a different reason — perhaps elevated AST/ALT — and the radiology report mentions increased hepatic iron. The patient is, somehow, iron-deficient by lab cutoffs and iron-loaded on imaging at the same time.
This is the iron paradox. Hemoglobin falls because iron isn’t getting to the bone marrow in usable form — the ferroxidase step that loads iron onto transferrin requires copper-bearing ceruloplasmin, and when ceruloplasmin is functionally low, iron piles up in storage tissues (liver, spleen, brain, heart, pancreas) instead of being shuttled into red-cell production. Ferritin in the bloodstream may read low because what little iron is moving is sequestered intracellularly as hemosiderin and as tissue ferritin that the standard serum ferritin assay does not capture. Meanwhile, supplemental iron poured into this broken system has nowhere to go but the wrong places — the very tissues that least need it — where it accelerates Fenton-reaction free-radical generation, deepens oxidative stress, drives more inflammation, and worsens fatigue. The patient is treated harder for “iron deficiency” while the underlying problem — a copper–ceruloplasmin lesion — goes untouched.
Robbins reframes the standard CBC and iron panel through this lens. Anemia in the chronically inflamed adult is rarely true storage-iron deficiency; it is an iron-handling failure. The corrective is to restore copper, magnesium, and retinol so ceruloplasmin can be made — not to throw more iron into a system that cannot use it.
2. Free Iron vs. Bound Iron
Iron biology is, at its heart, about containment. Healthy iron is sequestered. In plasma it is bound to transferrin (which delivers it to cells). Inside cells, what isn’t immediately used is locked into ferritin nanocages or built into heme proteins — hemoglobin, myoglobin, the cytochromes of the electron transport chain. Under normal conditions, less than 0.1% of total body iron exists as free or “labile” iron at any moment. The body works very hard to keep that fraction tiny.
The reason it works so hard is the Fenton reaction. Free Fe²⁺ reacts with hydrogen peroxide to generate the hydroxyl radical (·OH) — the most damaging reactive oxygen species in biology, capable of attacking DNA bases, lipids, and protein thiols indiscriminately. Even nanomolar concentrations of free iron in the wrong cellular compartment cause measurable oxidative damage. Ferroptosis — an iron-dependent form of regulated cell death distinct from apoptosis or necrosis — was characterized in the 2010s and has since been implicated in stroke, heart attack, neurodegeneration, and chemotherapy response. The take-home is simple: iron in the wrong place, at the wrong time, in the wrong oxidation state, is a poison.
Ceruloplasmin’s ferroxidase activity exists precisely to keep this from happening. By oxidizing Fe²⁺ back to Fe³⁺ for transferrin loading, ceruloplasmin keeps the labile-iron pool small and keeps iron moving in regulated channels. When ceruloplasmin function falls — whether through copper deficiency, retinol deficiency, magnesium-dependent enzyme failure, or rare aceruloplasminemia — the labile pool grows. The clinical picture is exactly what Robbins describes: oxidative stress, fatigue, inflammation, tissue iron accumulation, and a serum panel that looks confusingly “low iron” even as the cells are drowning in it.
3. Ferritin: Storage Marker or Inflammation Marker?
Ferritin is two things at once, and the failure to remember that is responsible for a great deal of misdiagnosis. Ferritin is, first, a real iron-storage protein — a 24-subunit nanocage that sequesters up to ~4,500 iron atoms per molecule inside cells. Serum ferritin (a small fraction of total ferritin, secreted by hepatocytes and macrophages) does correlate with body iron stores in healthy people. But ferritin is also, second, an acute-phase reactant — an inflammation-induced protein whose serum level rises in response to IL-6 and other inflammatory cytokines, independent of actual iron stores. In a patient with autoimmune disease, fatty liver, metabolic syndrome, chronic infection, or active inflammation of any origin, serum ferritin rises because the liver is making more of it as part of the host-defense program of sequestering iron away from invading pathogens. That ferritin reading no longer represents iron stores.
Standard reference ranges (typically 30–300 ng/mL for adults) reflect a population that is heavily inflamed by modern standards. Robbins, drawing on the Iron Disorders Institute’s work, argues that the healthy ferritin range is much narrower — 50–150 ng/mL — and that ferritin above ~100 ng/mL in a patient with any inflammatory condition is far more likely to be reading inflammation than reading iron. His clinical target is ferritin under 80 ng/mL in adult men and under 50 ng/mL in women. This is not actually radical: mainstream hepatology already adjusts the interpretation of elevated ferritin downward when CRP is elevated, and the literature on hyperferritinemia in metabolic syndrome (where ferritin is elevated without iron overload on liver biopsy) supports the inflammation-marker reading.
The mirror-image error is reading low ferritin in a non-bleeding adult as definitive iron deficiency. In a menstruating woman with heavy periods, low ferritin plus low transferrin saturation plus a low reticulocyte count truly is iron deficiency and may need iron. But in an adult man with fatigue, “low” ferritin (say, 25 ng/mL) without any blood loss, with normal hemoglobin, and with intact red-cell indices is much more likely to reflect functional copper deficiency — iron is being taken in normally but is not being mobilized into the storage pool because ferroxidase activity has failed. In Robbins’s framing, the fix is copper, not iron. The next test to order is a full mineral panel, including ceruloplasmin and serum copper, before reaching for the ferrous sulfate.
4. The Iron-Fortified American Food Supply
Most Americans dramatically underestimate how much iron they consume each day, because most of it is added without their knowledge. In 1941, the United States made iron fortification of refined wheat flour mandatory under the Enrichment Act, replacing the iron, niacin, riboflavin, thiamin, and folate that milling stripped from whole wheat. The current standard adds about 14 mg of iron per pound of flour — roughly 4 mg per typical four-ounce slice of bread. By the 1960s, ready-to-eat breakfast cereals had become a second mandatory delivery vehicle for iron; some popular brands deliver 18 mg per serving, a full 100% of the adult RDA, in a single bowl. Infant formulas are iron-fortified by design. Pasta is enriched. Rice is enriched. The bagel, the pizza crust, the breakfast sandwich, the kid’s cereal, the after-school crackers, and the dinner pasta are all delivering added inorganic iron that the body cannot easily refuse.
Add to this a typical American diet that includes red meat (rich in heme iron, which is absorbed unregulated, unlike non-heme iron), and add to that the routine use of multivitamins and prenatal vitamins containing 18–27 mg of iron, and the average adult is taking in 25–40 mg of iron per day. The physiological daily loss in a non-menstruating adult is closer to 1–2 mg. Even adjusting for low absorption efficiency of non-heme iron, the cumulative imbalance over decades is substantial. Several large epidemiologic correlations have linked rising national iron-fortification policies with rising rates of metabolic syndrome, type 2 diabetes, non-alcoholic fatty liver disease, and cardiovascular disease — correlations that are confounded but suggestive. Countries that fortify aggressively tend to have higher rates of these conditions; countries that do not fortify tend to have lower rates after controlling for diet quality.
Robbins’s practical advice is straightforward: read the ingredient panel of every packaged food. Look for the words iron, ferrous sulfate, ferrous gluconate, ferrous fumarate, ferric pyrophosphate, or reduced iron. If they appear, the food is delivering inorganic iron that does not require copper for absorption and bypasses the body’s normal iron-regulation machinery. For an adult attempting iron off-loading, this is the single largest controllable input.
5. Hereditary Hemochromatosis & The Spectrum
Classical hereditary hemochromatosis (HH) is the genetic disorder of uncontrolled iron absorption. The most common form is HFE-related: homozygosity for the C282Y mutation produces a defective HFE protein that fails to signal hepcidin properly, leaving the gut continuously absorbing iron as if the body were always deficient. The result, untreated, is progressive iron loading of the liver (cirrhosis, hepatocellular carcinoma), heart (cardiomyopathy, arrhythmia), pancreas (“bronze diabetes”), pituitary (hypogonadism), joints (arthropathy), and skin (the classic bronze pigmentation). The treatment is therapeutic phlebotomy — weekly to monthly blood removal until ferritin and transferrin saturation normalize, then maintenance phlebotomy two to four times a year for life.
What is less widely recognized is the breadth of the hemochromatosis spectrum. C282Y homozygotes are roughly 1 in 200 in populations of Northern European descent — the highest frequency of any clinically significant Mendelian disorder in those populations. C282Y heterozygotes (1 in 8 to 1 in 10 in the same populations) accumulate iron at lower rates but, in combination with other factors (alcohol intake, hepatitis C infection, metabolic syndrome), can develop clinically meaningful overload. The H63D mutation is even more common; C282Y/H63D compound heterozygotes have intermediate risk. Beyond HFE, juvenile hemochromatosis caused by hemojuvelin (HJV) or hepcidin (HAMP) mutations produces severe, early-onset overload. Mutations in transferrin receptor 2 (TfR2) and ferroportin (SLC40A1) cause additional forms. The aggregate prevalence of clinically meaningful iron-loading variants in European-descent populations is closer to 1 in 30 to 1 in 50 — not the rarity often portrayed.
Robbins extends this further. He argues that even non-HFE patients accumulate iron in the modern environment because the regulatory system was never designed for the iron load now being delivered. Whether or not one accepts that broader extension, the clinical implication is the same: any patient with persistently elevated ferritin, especially with a Northern European background, deserves an iron-panel workup including transferrin saturation and (if indicated) HFE genotyping. Iron loading is far more common than the textbooks suggest, and missing it has decade-long consequences.
6. Brain Iron Accumulation in Neurodegeneration
The brain handles iron with extraordinary care — and when that handling fails, the consequences are devastating. Iron is required for myelin synthesis, neurotransmitter production (dopamine synthesis depends on iron-containing tyrosine hydroxylase), and oxidative phosphorylation in mitochondria-rich neurons. But iron in the wrong neuronal compartment, in the wrong oxidation state, is catastrophic. The substantia nigra accumulates iron progressively in Parkinson’s disease, visible on T2*-weighted and R2* MRI sequences and correlating with disease severity. The hippocampus and cortex accumulate iron in Alzheimer’s, where it co-localizes with amyloid-β plaques. The basal ganglia accumulate iron in Friedreich’s ataxia and in pantothenate-kinase-associated neurodegeneration (PKAN). The cerebellum accumulates iron in some hemochromatosis variants.
The mechanism converges on the same chemistry: iron-driven Fenton-reaction free-radical generation, propagated by ferroptosis, in cells that already have high baseline oxidative metabolism and limited regenerative capacity. Iron also accelerates α-synuclein and amyloid-β aggregation in vitro, suggesting it is not just an innocent bystander but a co-pathogen. Iron disrupts the blood-brain barrier in animal models, allowing further peripheral inflammation and immune-cell infiltration into the parenchyma. The 2014 deferiprone trial in Parkinson’s disease (Devos et al.) was the first proof-of-concept that lowering brain iron with a chelator could produce modest clinical improvement, and follow-on trials are ongoing.
From the RCP perspective, the prevention story is more important than the treatment story. Brain iron accumulation does not start in the seventh decade — it accumulates over a lifetime of dietary iron loading and copper depletion. Anyone with a family history of neurodegenerative disease has a strong rationale to think about iron load decades before symptoms appear, and to think about ceruloplasmin function as the upstream protective variable. The brain has very limited iron off-loading capacity once accumulated; the lever that moves easily is the input side — what comes in via diet and supplements, and how well the ferroxidase system can keep iron sequestered.
7. Therapeutic Phlebotomy: Donating Blood as Medicine
Therapeutic phlebotomy is the most efficient iron-removal method available, by a wide margin. A single 500 mL whole-blood donation removes roughly 250 mg of iron — the iron contained in the hemoglobin of the removed red cells. Compare this to oral chelation, which removes perhaps 50–80 mg per day at substantial cost and side-effect burden. Three or four blood donations per year off-load 750–1,000 mg of iron, which can shift ferritin from the high triple digits down into the healthy 50–80 range over twelve to eighteen months in an adult man whose dietary inputs have also been reduced.
The American Red Cross requires an 8-week interval between whole-blood donations for men and 56 days for women, with hemoglobin thresholds at the time of donation. For patients with diagnosed hemochromatosis, formal therapeutic phlebotomy under medical supervision proceeds on a more aggressive schedule — weekly or biweekly during the de-iron phase, then maintenance every two to four months. A standing order from a treating physician converts what would otherwise be a regular blood donation into a coded therapeutic procedure, often covered by insurance.
For the broader Robbins audience — adults without classical hemochromatosis but with elevated ferritin, metabolic syndrome, fatty liver, or family history of neurodegenerative disease — routine blood donation through the Red Cross or a local blood bank is a no-cost, well-tolerated, and powerful iron-management tool. Eligibility, screening, and aftercare are all standardized. Anyone with elevated ferritin and no contraindication can use it. The cost-benefit ratio is unusually favorable: donate two to four times per year, lower body iron stores, contribute to the blood supply, and check ferritin annually to monitor trajectory.
8. When Iron Supplementation IS Genuinely Needed
Robbins is sometimes mischaracterized as an absolute opponent of iron supplementation. He is not. The RCP framework recognizes specific clinical situations where iron supplementation is both appropriate and essential:
- Active gastrointestinal bleeding — visible bleeding (melena, hematochezia, hematemesis) or occult bleeding identified by stool testing. Iron loss exceeds replacement and supplementation is required while the source is identified and treated.
- Heavy menstrual losses with documented true iron deficiency — all three of low ferritin, low transferrin saturation, and low reticulocyte count, ideally with low retic-Hb content. Heavy menstrual bleeding is the most common cause of true iron-deficiency anemia in adult women.
- Pregnancy with confirmed deficiency — expanded blood volume and fetal demand can outpace dietary intake even in well-nourished women. Confirmed deficiency on labs warrants supplementation.
- Post-bariatric surgery malabsorption — gastric bypass patients lose acid-dependent iron absorption and frequently need supplemental iron lifelong.
- Iron-deficient infants — rare in iron-fortified countries, common in low-income global settings where the public-health calculus is different. The WHO supplementation programs exist for clear reasons in those populations.
Even in these legitimate cases, Robbins would emphasize co-supplementation with copper, retinol (vitamin A), and the cofactors needed for ceruloplasmin synthesis so that the supplemental iron can actually be loaded and used safely. He would prefer food-form iron — red meat, organ meats (especially beef liver), blackstrap molasses — over isolated ferrous-salt tablets, because food-form iron arrives packaged with the cofactors that help direct it to the right places. And he would re-test in three to four months and de-escalate as soon as the deficiency is corrected, rather than continuing supplementation indefinitely.
9. Practical Approach to Iron Off-Loading
The RCP iron off-loading protocol is layered — reduce inputs, increase outputs, and restore the regulatory machinery in parallel.
- Stop ADDED iron. Read every label. Remove iron-fortified breakfast cereals, multivitamins with iron, prenatal vitamins (if not pregnant), iron-containing protein powders, and iron-fortified breads where possible. Choose unfortified whole-grain or sourdough bread, oatmeal, and whole-food breakfasts (eggs, fruit, real cheese) instead of fortified cereal.
- Donate blood every 8 to 16 weeks for men, or after menses cease for women. Check ferritin annually to monitor progress. Document the trajectory.
- Drink coffee with iron-containing meals. Chlorogenic acid in coffee reduces non-heme iron absorption by roughly 60%. A morning coffee with breakfast, or coffee within an hour of a meal, blunts iron uptake meaningfully without requiring any other change.
- Drink black or green tea with meals. Tea tannins reduce iron absorption similarly to coffee polyphenols. Tea with lunch or dinner, every day, is a passive iron-reduction lever.
- Calcium with meals (food calcium, not supplemental). Calcium competes with iron for absorption at the brush-border level. A glass of milk, a serving of yogurt, or sardines (calcium plus omega-3) at the same meal as iron-containing foods reduces uptake. Note that this conflicts with the RCP’s general recommendation to stop calcium supplements, so use food calcium specifically — dairy, sardines with bones, leafy greens.
- Separate vitamin C from iron-containing meals. High-dose ascorbic acid increases non-heme iron absorption substantially — the opposite of what the iron-loaded patient wants. The RCP already discourages synthetic ascorbic acid for other reasons; this is an additional one. If using whole-food vitamin C (acerola, camu camu), take it on an empty stomach, away from iron-containing meals.
- Restore ceruloplasmin. The off-loading effort is incomplete without this. Bioavailable copper from beef liver, oysters, and cacao; retinol from cod-liver oil and liver; magnesium glycinate; whole-food vitamin C; and removal of the depleting inputs (fluoride, glyphosate, synthetic D3 at high doses) are the inputs to ceruloplasmin synthesis. See the Ceruloplasmin and Whole-Food Copper articles for details.
- Re-test ferritin every four to six months. Track the trajectory. Adjust phlebotomy frequency, food-iron intake, and ceruloplasmin support based on the numbers. Aim for ferritin in the healthy range — 50–80 ng/mL for adult men, lower for women — with a stable transferrin saturation of 20–35%.
This protocol is not aggressive. It is patient. Iron took years or decades to load, and it will take twelve to twenty-four months of consistent off-loading to bring stores into the healthy range while simultaneously rebuilding the ferroxidase system. For most adults, the changes are tolerable — many were going to drink the coffee, donate the blood, and skip the fortified cereal anyway. What changes is the framing: these are not random lifestyle choices, they are an iron-management system, and they are the upstream lever for a great deal of metabolic, cardiovascular, and neurological resilience over a lifetime.
10. Key Research Papers
- Hereditary hemochromatosis (HFE)
- Ferritin as an acute-phase reactant
- Brain iron in neurodegeneration (MRI)
- Parkinson’s disease & substantia nigra iron
- Alzheimer’s, iron & amyloid
- Therapeutic phlebotomy & ferritin
- Iron fortification & disease risk
- Non-HFE hemochromatosis
- Fenton reaction, iron & ROS
- Deferiprone in Parkinson’s disease
- Hepcidin & iron regulation
- Ferroptosis: iron-dependent cell death
Connections
- Morley Robbins
- Ceruloplasmin and Bioavailable Copper
- Relationship Between Hemoglobin and Ceruloplasmin — why ceruloplasmin (the copper ferroxidase) is required to load iron into hemoglobin, and why "iron-deficiency anemia" is often functional copper deficiency.
- Whole Food Copper Sources
- Copper-Iron Dysregulation
- Root Cause Protocol
- Magnesium Replenishment
- Vitamin D Controversy
- Adrenal Cortisol and Mineral Connection
- Cure Your Fatigue
- Iron
- Copper
- Magnesium
- Hemochromatosis
- Berberine and Iron Overload
- Liver Cleansing
- Iron Panel
- Metabolic Syndrome
- Glyphosate Copper Chelation